跟着Molecular Plant学作图:R语言circlize包画圈图展示基因组的一些特征
跟着Molecular Plant学作图:R语言circlize包画圈图展示基因组的一些特征

用户7010445
发布于 2023-01-06 12:15:46
发布于 2023-01-06 12:15:46
代码可运行
运行总次数:2
代码可运行
论文
A telomere-to-telomere gap-free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding
https://www.cell.com/molecular-plant/fulltext/S1674-2052(22)00192-7?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1674205222001927%3Fshowall%3Dtrue
今天的推文我们重复一下论文中的Figure1a

image.png
论文中没有提供数据和代码,数据自己算,代码自己写
之前分享过的关于圈图的推文
跟着Nature Communications学画图:R语言circlize包画弦图展示基因密度
计算gc含量和基因密度
利用基因组fasta文件统计染色体长度和GC含量,自己写python脚本(当然有很多工具可以统计)利用gtf文件统计基因密度
读取fasta用到了biopython
class WaterMelon:
def __init__(self,gtf,fasta):
self.gtf = gtf
self.fasta = fasta
def chromoLen(self):
chrLen = {}
for rec in SeqIO.parse(self.fasta,'fasta'):
chrLen[rec.id] = len(rec.seq)
return chrLen
def gcContent(self,gc_window):
self.gc_window = gc_window
gc_content = {'chr_id':[],
'bin_start':[],
'gc':[]}
chr_len = self.chromoLen()
#print(chr_len)
for rec in SeqIO.parse(self.fasta,'fasta'):
for i in range(0,chr_len[rec.id],self.gc_window):
#print(rec.id)
gc_content['chr_id'].append(rec.id)
gc_content['bin_start'].append(i)
gc_content['gc'].append(round(GC(rec.seq[i:i+self.gc_window]),2))
return pd.DataFrame(gc_content)
def geneDensity(self,gene_window):
self.gene_window = gene_window
final_df = []
df = pd.read_table(self.gtf,header=None,comment="#",sep="\t",
usecols=[0,2,3,4],
names="Chromosome Feature Start End".split())
#df.columns = "Chromosome Source Feature Start End Score Strand Frame Attribute".split()
df = df[df.Feature=="gene"]
chrLen = self.chromoLen()
for chr_id in chrLen.keys():
print(chr_id)
df1 = df[df.Chromosome==chr_id]
gene_start = [int(a) for a in df1.Start]
gene_start.insert(0,0)
gene_start.append(round(chrLen[chr_id]/self.gene_window)*self.gene_window+self.gene_window)
#print(gene_start)
bin_start = [int(a.left) for a in pd.cut(gene_start,bins=round(chrLen[chr_id]/self.gene_window)+1).value_counts().index]
bin_start[0] = 0
gene_count = list(pd.cut(gene_start,bins=round(chrLen[chr_id]/self.gene_window)+1).value_counts().values)
#print(len(bin_start))
#print(len(gene_count))
#print("OK")
final_df.append(pd.DataFrame({'chr_id':chr_id,'bin_start':bin_start,'gene_count':gene_count}))
return pd.concat(final_df)
这里是用python算的
TE的密度暂时不知道怎么算,snp和indel的密度可以使用vcftools软件,但是没有找到输入文件
首先是读取数据
df<-read.csv("D:/Jupyter/bioinformatics_python/MPwatermelon.csv")
gc<-read.csv("D:/Jupyter/bioinformatics_python/gc.csv")
genedensity<-read.csv("D:/Jupyter/bioinformatics_python/genedensity.csv")
max(df$chr_len)
head(gc)
range(gc$gc)
head(genedensity)
range(genedensity$gene_count)
genedensity %>%
mutate(group=case_when(
gene_count < 35 ~ "#1efc05",
gene_count > 50 ~ "red",
TRUE ~ "black"
)) -> genedensity
染色体长度数据

image.png
gc含量数据

image.png
基因密度数据
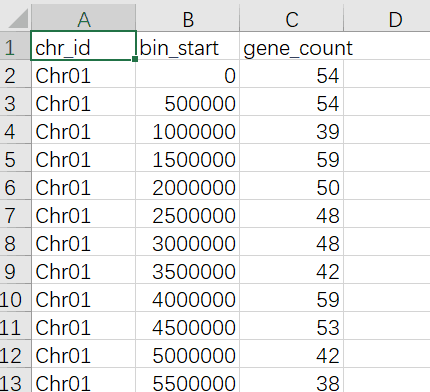
image.png
作图代码
library(circlize)
brk <- seq(0,40,by=2)*10^6
brk.label<-c()
for (i in brk){
ifelse(i%%10^7==0,brk.label<-append(brk.label,
paste0(i/10^7,"0M")),
brk.label<-append(brk.label,""))
}
brk.label[1]<-"0M"
brk.label
#circos.par(points.overflow.warning = FALSE)
pdf(file = "mp.pdf",
width = 15,height = 15)
circos.par(start.degree =86,clock.wise = T)
circos.initialize(factors = df$chr_id,
xlim = matrix(c(rep(0,11),df$chr_len),ncol=2))
circos.trackPlotRegion(df$chr_id,
ylim = c(0, 10),
track.height = 0.1,
bg.border = NA,
#ylim=CELL_META$ylim,
panel.fun = function(x, y) {
circos.text(mean(CELL_META$xlim), 12,
get.cell.meta.data("sector.index"))
})
circos.trackPlotRegion(df$chr_id,
ylim = c(0, 100),
track.height = 0.1,
bg.col = '#EEEEEE6E',
bg.border = NA)
for (chromosome in df$chr_id){
circos.axis(sector.index = chromosome,
h = 110,
major.at = brk,
minor.ticks = 0,
labels = brk.label,
labels.facing="clockwise",
labels.cex = 0.6)
}
circos.trackLines(gc$chr_id,gc$bin_start,gc$gc,
area = TRUE,
col = "red",
border="transparent")
circos.trackPlotRegion(df$chr_id,
ylim = c(0, 10),
track.height = 0.1,
bg.col = '#EEEEEE6E',
bg.border = NA)
for (chromosome in df$chr_id){
circos.segments(sector.index = chromosome,
x0=genedensity[genedensity$chr_id==chromosome,]$bin_start,
x1=genedensity[genedensity$chr_id==chromosome,]$bin_start,
y0=0,y1=10,col=genedensity[genedensity$chr_id==chromosome,]$group)
}
circos.trackPlotRegion(df$chr_id,
ylim = c(0, 10),
track.height = 0.1,
bg.col = '#EEEEEE6E',
bg.border = NA)
for (chromosome in df$chr_id){
circos.segments(sector.index = chromosome,
x0=genedensity[genedensity$chr_id==chromosome,]$bin_start,
x1=genedensity[genedensity$chr_id==chromosome,]$bin_start,
y0=0,y1=10,col=genedensity[genedensity$chr_id==chromosome,]$group)
}
circos.trackPlotRegion(df$chr_id,
ylim = c(0, 100),
track.height = 0.1,
bg.col = '#EEEEEE6E',
bg.border = NA)
for (chromosome in df$chr_id){
circos.barplot(sector.index = chromosome,
value = genedensity[genedensity$chr_id==chromosome,]$gene_count,
pos = genedensity[genedensity$chr_id==chromosome,]$bin_start,
col = "red",
bar_width = 500000,
border="transparent")
}
circos.trackPlotRegion(df$chr_id,
ylim = c(0, 100),
track.height = 0.1,
bg.col = '#EEEEEE6E',
bg.border = NA)
for (chromosome in df$chr_id){
circos.barplot(sector.index = chromosome,
value = genedensity[genedensity$chr_id==chromosome,]$gene_count,
pos = genedensity[genedensity$chr_id==chromosome,]$bin_start,
col = "blue",
bar_width = 500000,
border="transparent")
}
circos.clear()
dev.off()
最终结果
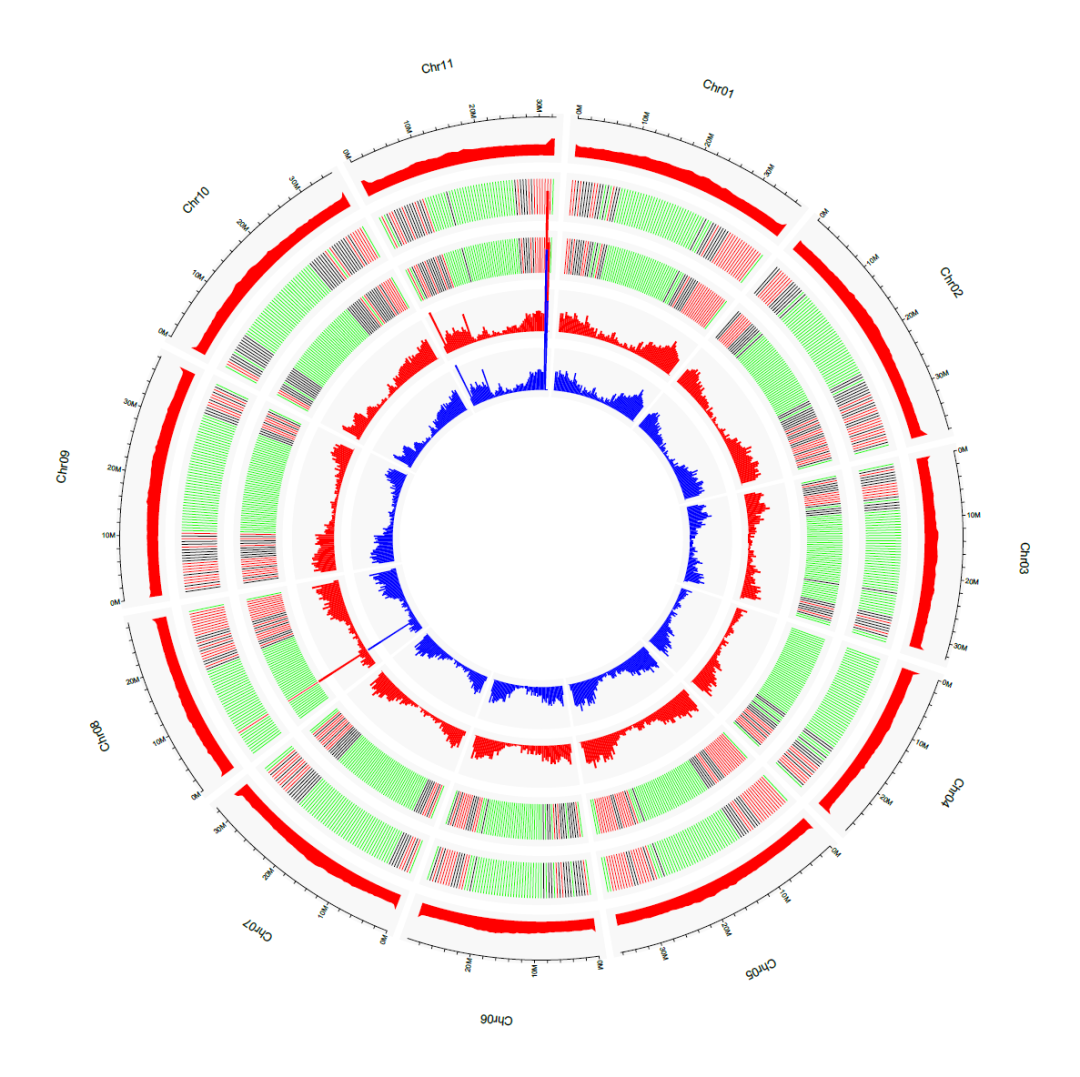
image.png
如何添加图例我暂时还没有搞明白,再好好学下这个包的用法,学会了再来分享
从外到内
- 第一圈是折线图,
- 第二、三圈是线段
- 第四五圈是柱形图
代码细节用文字描述可能会比较繁琐,抽时间录视频介绍吧
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2022-08-22,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
暂无评论
推荐阅读







推荐阅读
相关推荐
跟着Nature Communications学画图:R语言circlize包画弦图展示基因密度
更多 >
腾讯云开发者
