ChAMP甲基化芯片分析官方流程学习
原创

ChAMP(Chip Analysis Methylation Pipeline)是一个用于Illumina 450K和850K DNA甲基化芯片数据分析的R包。它是一个全面的工具包,可以从数据预处理到差异甲基化分析和功能注释提供一站式解决方案。它特别适用于甲基化数据的批处理分析和高通量研究。

- ChAMP 包专为分析 Illumina Methylation BeadArray 数据(包括 EPIC (860k芯片) 和 450k 芯片)而设计,提供了一整套整合当前 450k 和 EPIC 分析方法的工作流程。它支持多种不同的数据输入方式,例如来自 .idat 文件或 Beta 值矩阵的数据,并提供全面的质量控制分析工具(如质量控制图)。
- ChAMP 包含多种探针校正方法,包括 SWAN(Type-2 探针校正方法)、基于峰值的校正(PBC)和 BMIQ(默认选择)。
- 此外,ChAMP 集成了 minfi 包中广泛使用的功能归一化(Functional Normalization, FN)方法,并支持奇异值分解(Singular Value Decomposition, SVD)以深入分析批次效应。此外,ChAMP 使用 ComBat 方法校正多种批次效应,并可通过 RefbaseEWAS 调整细胞类型的异质性。
- ChAMP 还包含推断拷贝数变异(CNV)的功能,可用于 450k 和 EPIC 数据的分析。在差异甲基化区域(DMR)的检测中,ChAMP 提供了一种新的 Probe Lasso 方法,此外还保留了传统的 DMR 检测方法(如 Bumphunter 和 DMRcate)。对于需要检测差异甲基化区块(Differentially Methylated Blocks, DMB)的用户,ChAMP 的新版本引入了专门的功能来支持这一分析。
- 基因集合富集分析(Gene Set Enrichment Analysis, GSEA)也可以通过 ChAMP 实现,新版本中还加入了校正探针分布不均导致偏倚的方法。此外,新版本的 ChAMP 集成了 FEM 包,该包可以在用户定义的基因网络模块中推断不同表型之间的差异甲基化基因。
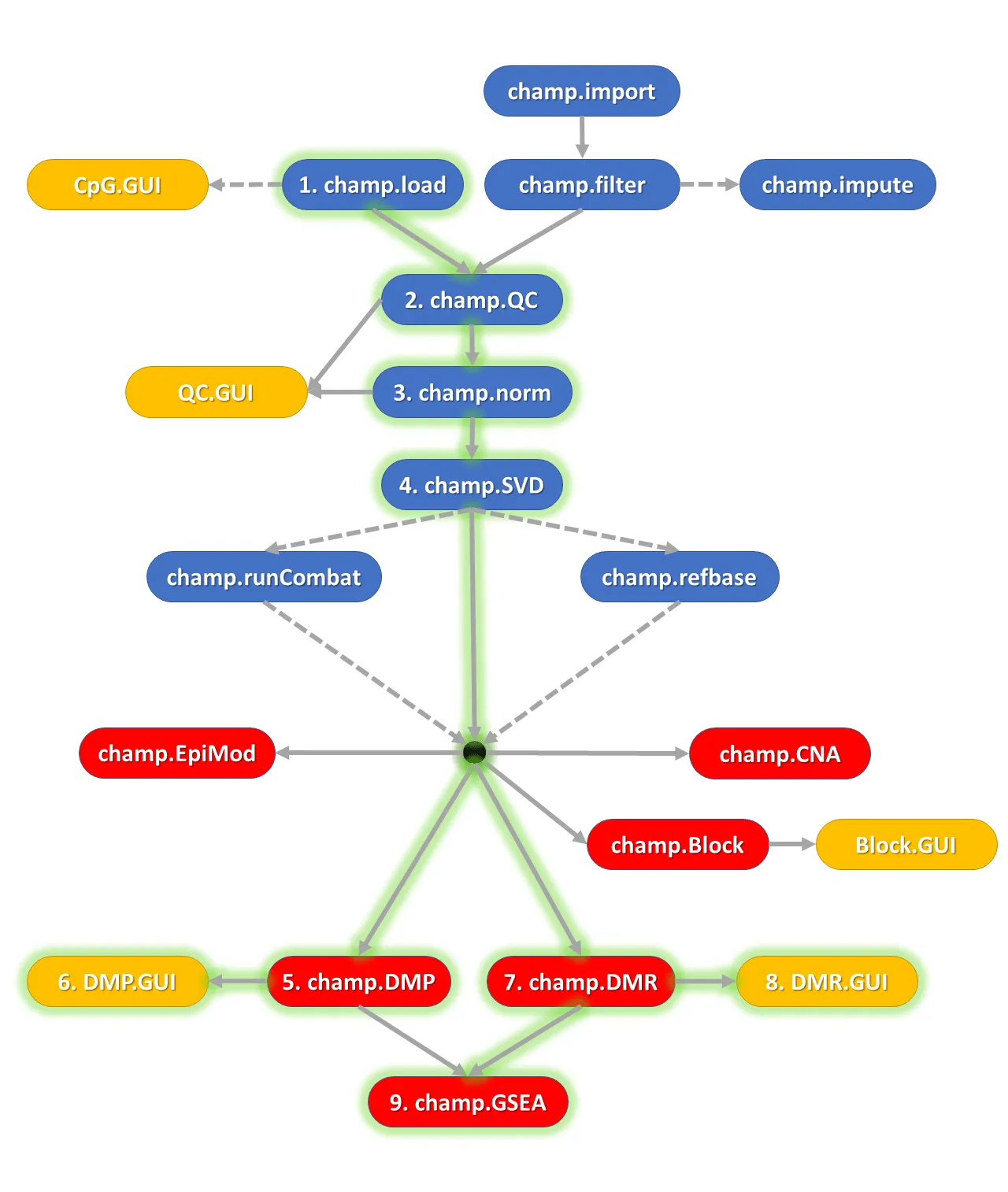
- 蓝色功能模块主要用于甲基化数据的预处理,包括数据加载、标准化、质量控制检查等步骤。
- 红色功能模块用于生成分析结果,包括差异甲基化位点(DMPs)、差异甲基化区域(DMRs)、差异甲基化区块(Differentially Methylated Blocks)以及通过 FEM 包衍生的差异甲基化基因模块检测方法(EpiMod,R包作者说目前不可用)和通路富集分析结果等。
- 黄色功能模块提供数据集和分析结果的图形用户界面(GUI)交互功能,便于用户直观地操作和查看分析结果。
目前ChAMP不仅可以分析原始.idat文件,也可以仅对甲基化beta矩阵和相应的表型进行分析。
分析步骤
1.导入
ChAMP安装可能会有很多问题,不过耐心安装就能解决。
rm(list = ls())
options(stringsAsFactors = F)
library("ChAMP")2.官方测试数据
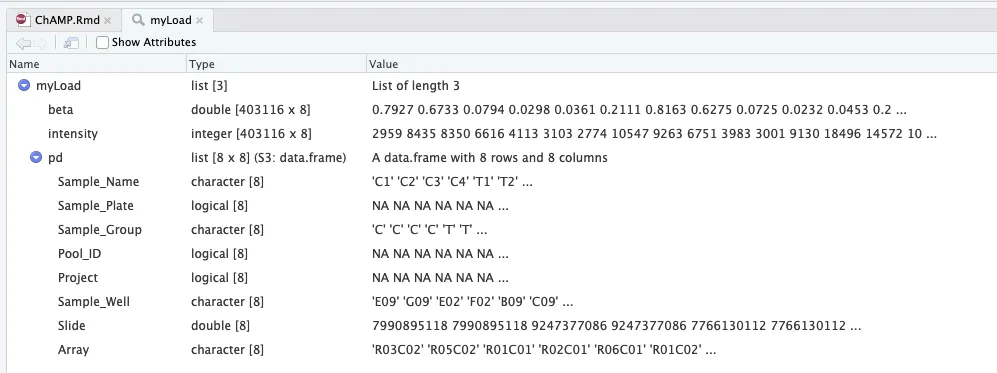
环境中增加5个数据:Anno,hm450.manifest.hg19, multi.hit, myLoad, probe.features
# 450k 肺肿瘤数据集包含8个样本,4个肺肿瘤样本(T)和4个对照样本(C)。
testDir=system.file("extdata",package="ChAMPdata")
myLoad <- champ.load(testDir,arraytype="450K")
# EPIC仿真数据
# data(EPICSimData)其中myLoad文件整合了beta值和表型信息,其中表型信息的列名是需要借鉴的,因为后续我们可能需要按照这个命名方式来创建自己数据的表型文件。
3.Full Pipeline
完整的流程可以通过一个命令运行(基本不会这么用的。。)
champ.process(directory = testDir)4.分开步骤
# filter中不需要输入内容,应该是自动检测了是否存在全局变量myImport?
myImport <- champ.import(testDir)
myLoad <- champ.filter()- 首先,过滤检测p值(默认值 > 0.01)超标的探针。检测p值存储在.idat文件中,champ.import会读取这些信息并将其转换为数据框。如果某个探针的p值高于0.01,则认为该探针失败(failed probe)。系统会在屏幕上输出一个failedSamples结果,显示每个样本中失败探针的比例。如果某些样本的失败探针比例较高(> 0.1),建议从分析中移除该样本并重新运行。研究发现,在许多情况下,大数据集中仅有一到两个样本因不合格而被排除,这些样本可能有70%甚至80%的探针失败。如果仅对探针进行过滤,则约80%的探针会被屏蔽。因此,ChAMP开发了一个新的过滤系统,用于同时评估样本和探针质量。如果某个样本的失败探针比例超过特定阈值(默认值=0.1),则会移除该样本,并对剩余样本进行探针过滤。样本和探针的阈值可通过参数SampleCutoff和ProbeCutoff分别控制。
- 其次,ChAMP 会过滤在至少5%的样本中具有少于3个beads的探针。此默认设置可通过参数filterBeads更改,或者通过参数beadCutoff调整频率。
- 第三,ChAMP 默认会过滤所有非CpG探针。
- 第四,默认情况下,ChAMP 会过滤所有与SNP相关的探针。SNP列表来源于Zhou等人在2016年发表的《Nucleic Acids Research》文章。如果已知数据来源的人群,可通过设置population参数选择特定人群进行过滤;否则,ChAMP 将使用Zhou提供的一般推荐探针列表进行过滤。
- 第五,默认设置下,ChAMP 会过滤所有多位点探针(multi-hit probes)。多位点探针列表来源于Nordlund等人在2013年发表的《Genome Biology》文章。
- 第六,ChAMP 会过滤位于X染色体和Y染色体上的探针。这也是默认设置,但用户可以通过参数filterXY更改。
以上所有过滤步骤都可通过champ.load和champ.filter函数中的参数控制,用户可以根据需求进行调整。需要注意的是,尽管大多数ChAMP功能支持独立的beta矩阵分析(不依赖于.idat文件),但champ.load不能单独对beta矩阵进行过滤。如果用户仅有beta矩阵和Sample_Sheet.csv文件,可以使用champ.filter函数执行过滤,并在后续使用其他函数进行分析。
质量控制
CpG.GUI(CpG=rownames(myLoad$beta),arraytype="450K")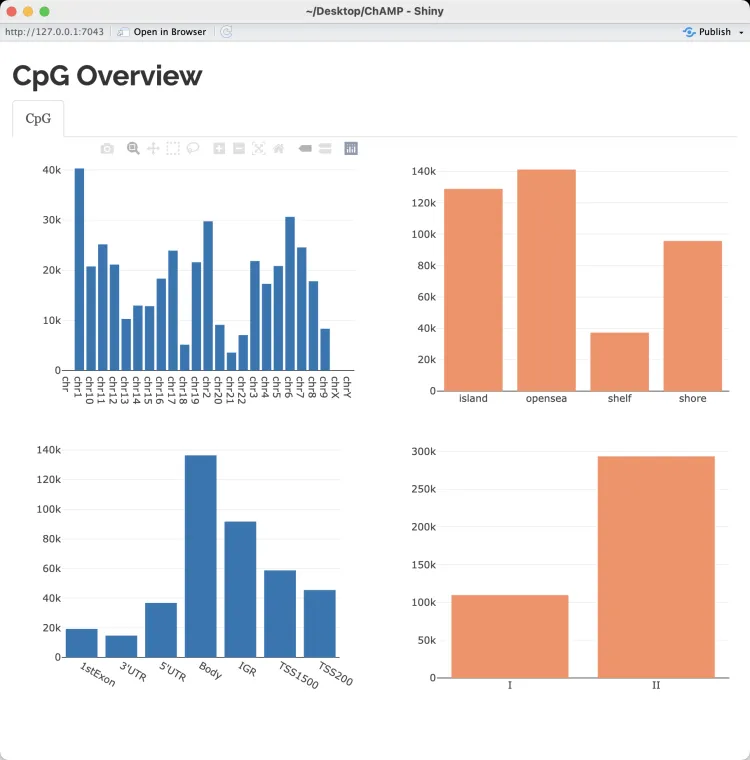
- 左上图(染色体分布):横轴表示染色体(chr1、chr2……chrX、chrY),纵轴表示在每条染色体上分布的 CpG 探针数量。
- 右上图(CpG 岛关系分布):横轴表示 CpG 探针与 CpG 岛的关系,包括“island”(CpG 岛内)、“opensea”(远离 CpG 岛)、“shelf”(CpG 岛边缘区域)和“shore”(CpG 岛附近区域)。纵轴表示各类别的探针数量。结果显示,绝大多数探针分布在 opensea 和 island 中,较少探针位于 shelf 和 shore。这与 CpG 探针的设计策略一致,大多数探针优先覆盖 CpG 岛及远离 CpG 岛的区域。
- 左下图(基因组区域分布):横轴表示探针在基因组中的位置,包括 1stExon(首个外显子)、3’UTR、5’UTR、Body(基因体区域)、IGR(基因间区域)、TSS1500(启动子区域,距转录起始位点1500bp以内)、TSS200(距转录起始位点200bp以内)。纵轴表示各区域的探针数量。从图中可以看出,绝大部分探针位于基因体(Body)区域,其次是 IGR 和启动子区域(TSS1500、TSS200)。这符合 CpG 探针设计的广泛覆盖特点。
- 右下图(探针设计类型分布):横轴表示探针的设计类型(I 型和 II 型),纵轴表示不同设计类型探针的数量。从图中可以看出,II 型探针的数量远高于 I 型探针。这是由于 Illumina 的甲基化芯片中,II 型探针的覆盖范围更广。
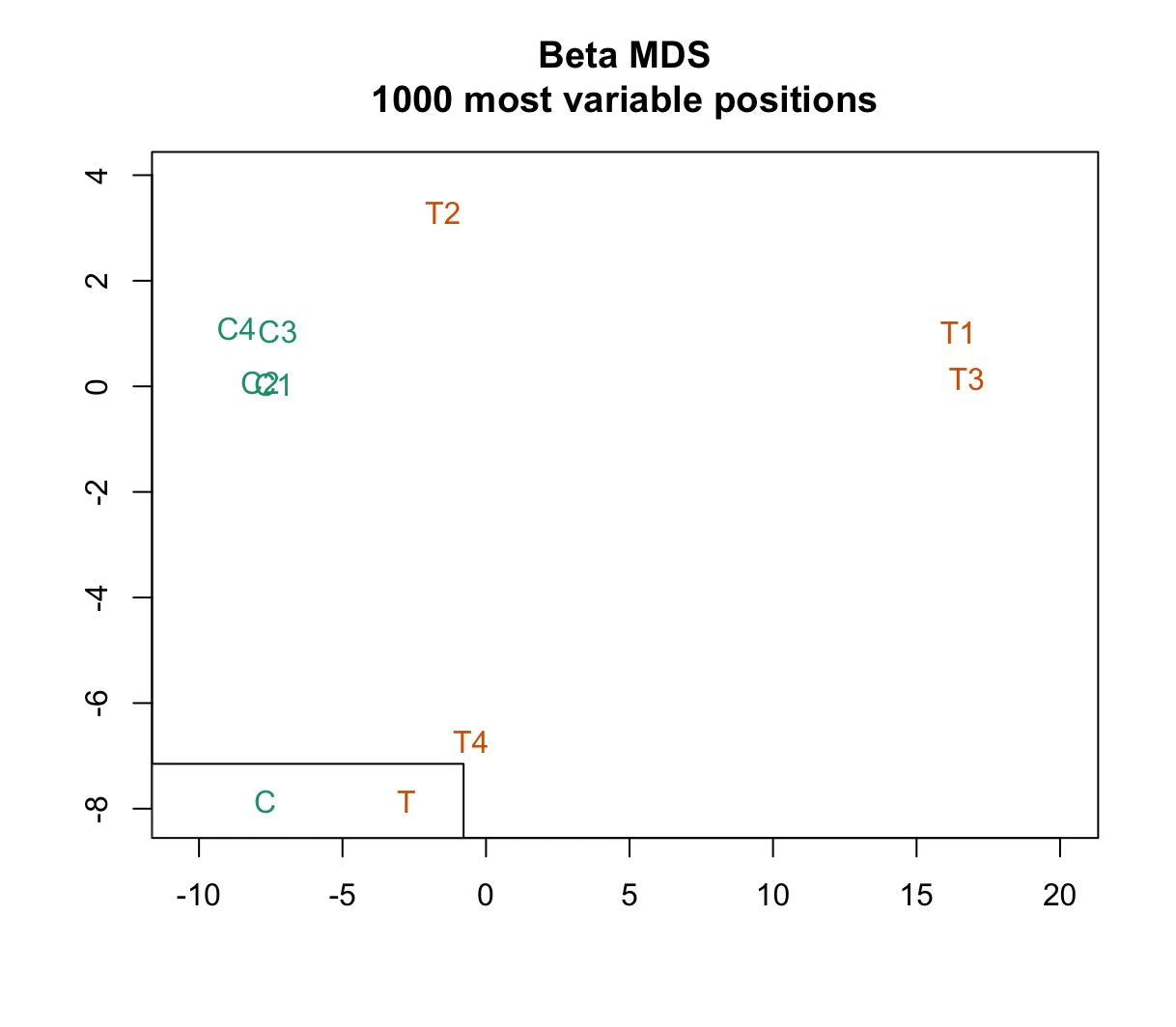
champ.QC(Feature.sel="None") # None, SVDmdsPlot:该图基于样本间最具变异性的1000个探针,直观地展示了样本之间的相似性。在图中,样本根据分组(Sample Groups)进行着色。
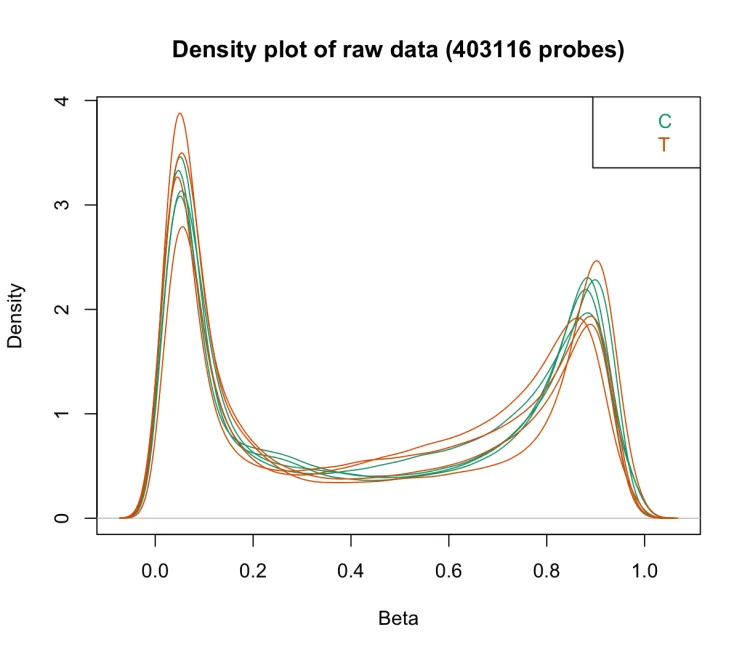
densityPlot:显示每个样本的Beta值分布;可以利用该图识别显著偏离其他样本的样本,这些样本可能质量不佳(如亚硫酸盐转化不完全)。此外,该图还可用于识别和确认甲基化或未甲基化的对照样本(如果研究中包含这些样本)。
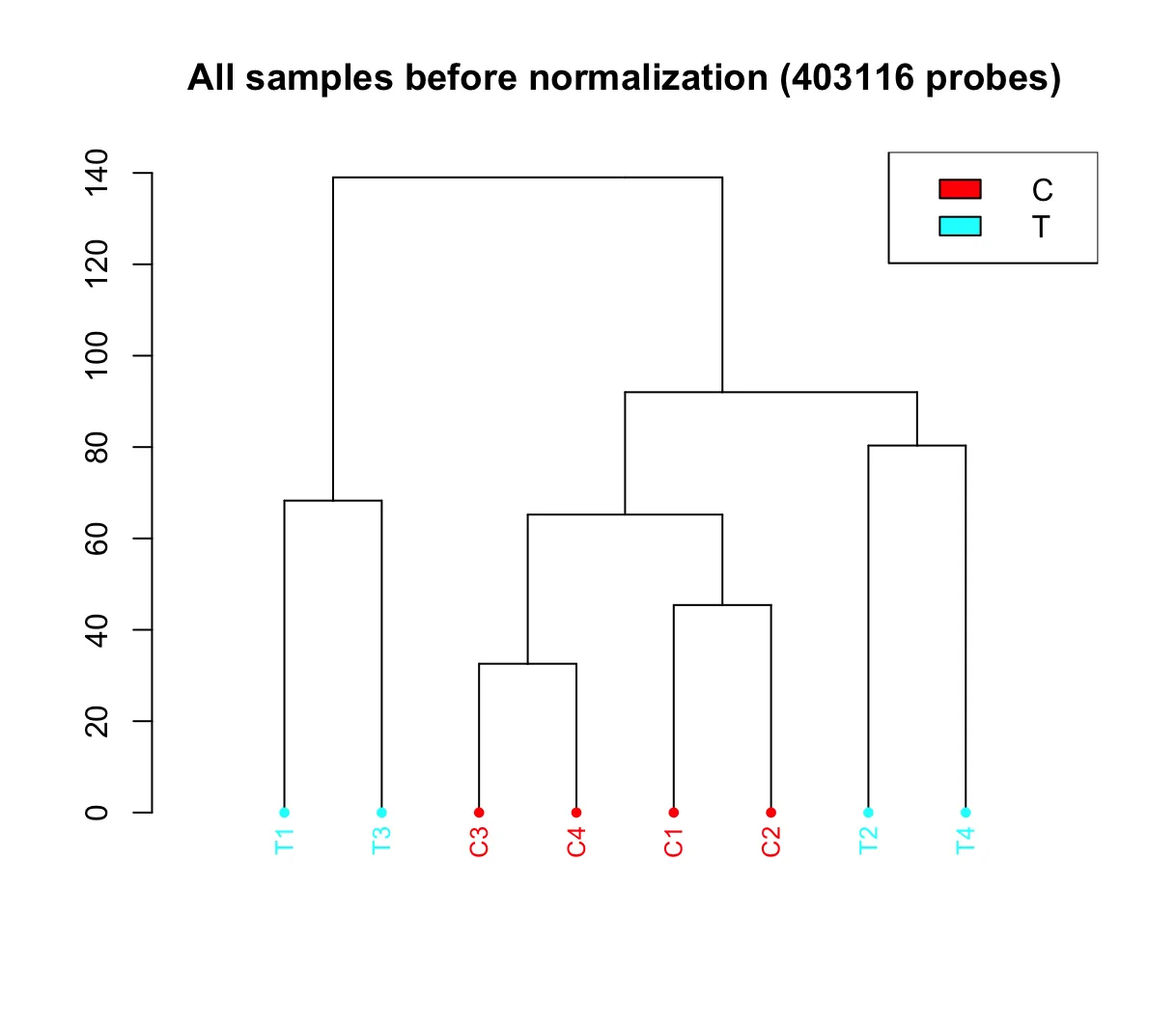
dendrogram:用于展示所有样本的聚类结果。可以选择不同的方法生成此图。在 champ.QC() 函数中,有一个参数 Feature.sel="None"。当参数设为“None”时,样本间的距离将直接基于所有探针计算(若数据集较大,可能导致服务器崩溃)。设为“SVD”时,champ.QC() 函数会采用 SVD 方法对 Beta 矩阵进行分解,并利用 “isva” 包中的 EstDimRMT() 方法选择显著成分,然后基于这些成分计算样本间的距离。
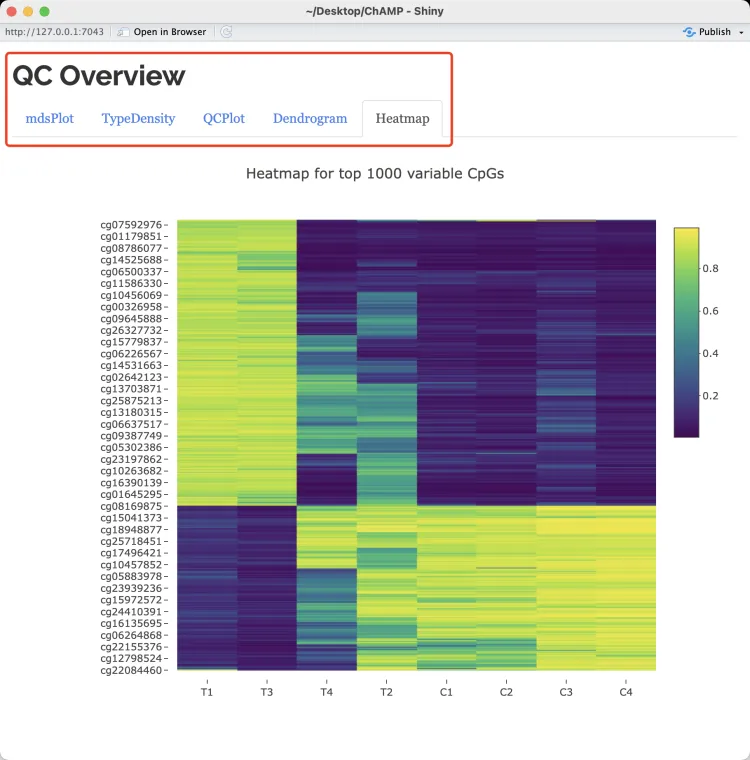
QC.GUI(beta=myLoad$beta,arraytype="450K")QC.GUI可以产生交互式界面,需要配置较高的电脑。
归一化(Normalize)
myNorm <- champ.norm(beta=myLoad$beta,arraytype="450K",cores=5)在Illumina BeadArray技术中,探针具有两种不同的设计类型(分别称为type-I和type-II),其杂交化学性质存在差异,这导致这两种探针的分布特性也不同。这是一种技术效应,与type-I和type-II探针因生物特性差异(如CpG密度)引起的变异无关。type-I和type-II探针甲基化分布的主要区别在于,type-II探针的动态范围较小。在监督分析中,这可能导致对type-I探针的偏好性选择。对于DMR(差异甲基化区域)检测,由于type-I和type-II探针可能同时位于相同的区域,校正这一偏差至关重要。因此,建议对type-II探针偏差进行校正,用户可以使用champ.norm函数执行此标准化。
- 在champ.norm函数中,提供了四种用于校正type-II探针偏差的方法:BMIQ、SWAN、PBC和FunctionalNormalization。这些方法之间存在关键差异,用户可参考相关文献选择最适合其分析需求的方法。
- 需要注意的是,FunctionalNormalization方法需要rgSet对象,SWAN方法需要mset和rgSet对象,这些对象需从IDAT文件中生成(如minfi包说明所述)。因此,FunctionalNormalization和SWAN标准化方法仅适用于采用“minfi”加载方法的分析。
- 值得注意的是,BMIQ功能已更新至1.6版本,为EPIC芯片数据提供了更好的标准化能力。然而,BMIQ可能无法收敛并输出结果,尤其是当样本的甲基化分布显著偏离三状态贝塔混合分布时(如在甲基化/非甲基化对照中可能发生),或样本质量较差时。此外,BMIQ函数现已支持并行运行,若计算机具有多个核心,用户可通过设置“cores”参数来加速运算。若设置PDFplot=TRUE参数,BMIQ函数生成的图形将保存至resultsDir目录。
奇异值分解
champ.SVD(beta = as.data.frame(myNorm), pd = myLoad$pd)
# If Batch detected, run champ.runCombat() here.
# myCombat <- champ.runCombat(beta=myNorm,pd=myLoad$pd,batchname=c("Slide"))Teschendorff提出的用于甲基化数据的奇异值分解方法(SVD)是一种强大的工具,可用于评估数据集中显著变异成分的数量和性质。这些变异成分理想情况下应与感兴趣的生物学因素相关,但通常也会与技术性变异来源(例如批次效应)相关。建议使用者尽可能收集分析样本的相关信息(例如日期、样本采集的季节、流行病学信息等),并在与SVD成分关联时纳入所有这些因素。如果样本是从.idat文件加载的,那么在champ.SVD()函数中设置参数RGEffect=TRUE时,BeadChip的18个内部探针控制(包括重亚硫酸盐转化效率)将被包括在内。 与旧版本ChAMP包相比,当前版本的champ.SVD()会检测所有有效的协变量进行分析,这意味着生成的图形会包含以下两种情况:
- 协变量的名称不能为name、Sample_Name或File_Name;
- 协变量必须包含至少两个值(例如,“BeadChip ID”作为协变量被测试时,研究中必须包含来自至少两个不同BeadChip的样本)。 champ.SVD()函数会自动检测pd文件中的所有有效协变量。因此,如果用户有一些希望被分析的独特协变量,可以将它们合并到pd文件中,从而允许champ.SVD()测试数据集中最显著变异成分与这些协变量的关联性。需要注意的是,champ.SVD()只能使用pd文件格式的表型数据。因此,如果用户有自己定义的协变量需要与当前pd文件一起分析,可以将协变量与myLoad$pd结合(通过cbind()),然后将此对象作为champ.SVD()函数pd参数的输入。
需要特别注意,不同类型的协变量(分类变量和数值型变量)在champ.SVD()中会使用不同的方法计算协变量与成分之间的显著性(分别为Krustal检验和线性回归)。因此,请确保pd对象是一个数据框,并将数值型协变量转换为“numeric”类型,将分类协变量转换为“factor”或“character”类型。如果协变量“Age”被指定为“character”类型,champ.SVD()将无法识别其应作为数值型变量进行分析。因此,我们建议用户对其数据集和pd文件有清晰的理解。
在champ.SVD()运行过程中,所有检测到的协变量都会打印在屏幕上,以便用户检查希望分析的协变量是否正确。结果将以热图形式显示(保存为SVDsummary.pdf),展示与提供的协变量信息相关的前几个主成分。在champ.SVD()中,使用isva包中的随机矩阵理论(Random Matrix Theory)检测潜变量的数量。如果检测到的成分数超过20,仅选择前20个主成分。在热图中,颜色越深表示p值越显著,表明SVD成分与感兴趣因素的关联越强。如果SVD分析显示技术因素占了很大比例的变异,则需实施其他归一化方法(例如ComBat)以帮助去除这些技术性变异。ComBat方法已包含在ChAMP流程中,可用于去除与BeadChip、位置和/或板块相关的变异,也可用于去除SVD分析中揭示的其他批次效应。
注意:在ChAMP的最新版本中,在champ.SVD()中增加了scree图,以帮助用户检查每个成分捕获的方差。用户可以根据捕获的方差确定批次效应的处理。例如,如果在降解分析后,前两个成分捕获了超过80%的方差,则无需关心后续成分中的批次效应,只需关注前两个成分中的显著关联即可。

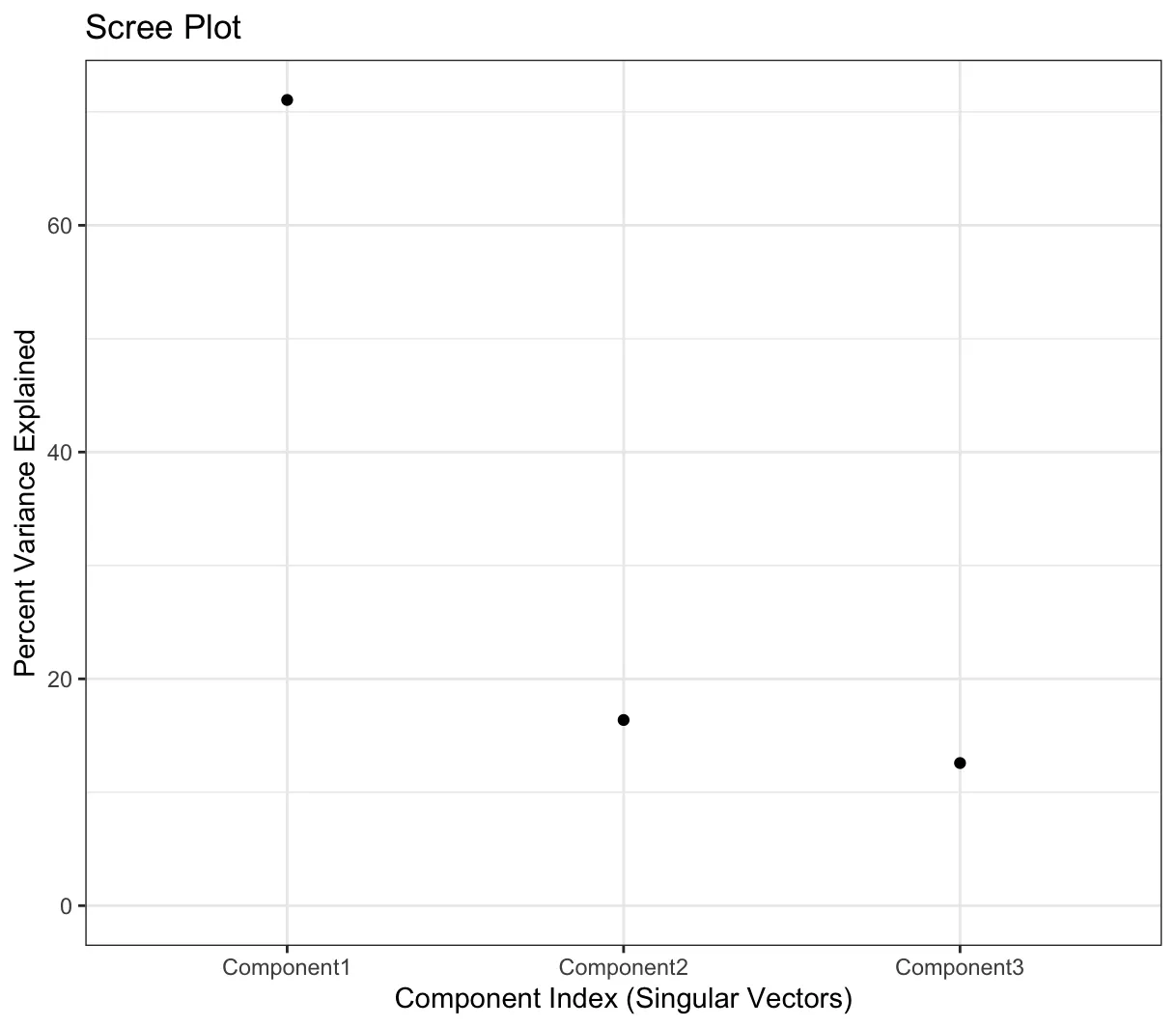
以上两个图片的结果有助于使用者了解数据的变异来源,比如上述内容显示是PC1占70%+的贡献,其中Sample_Group是最大的变异来源。
差异甲基化探针
# Differential Methylation Probes
myDMP <- champ.DMP(beta = myNorm,pheno=myLoad$pd$Sample_Group)
head(myDMP[[1]])
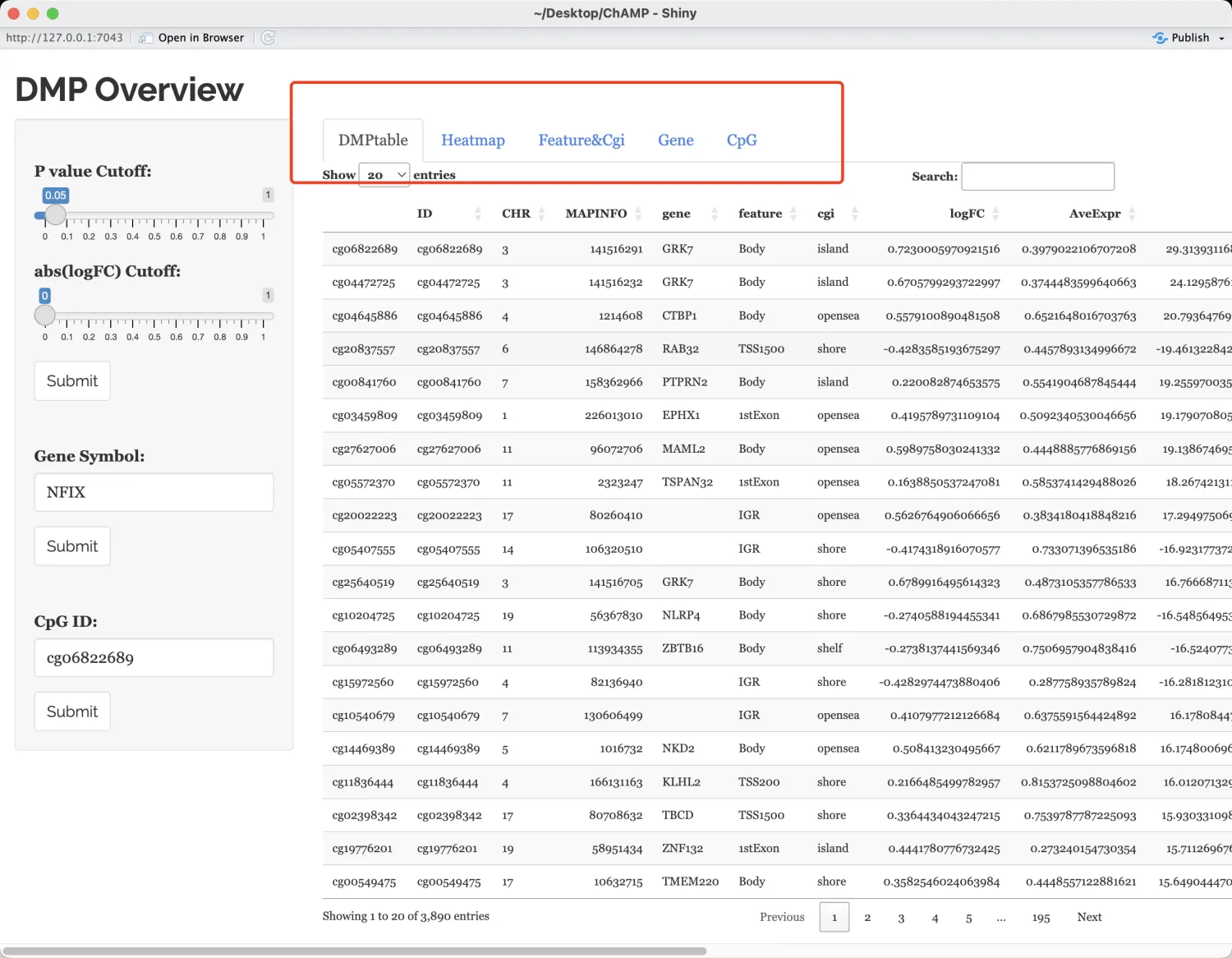
DMP.GUI(DMP=myDMP[[1]],beta=myNorm,pheno=myLoad$pd$Sample_Group)
DMP.GUI()DMPtable中可以输出详细的limma差异分析结果信息。
heatmap可视化展示
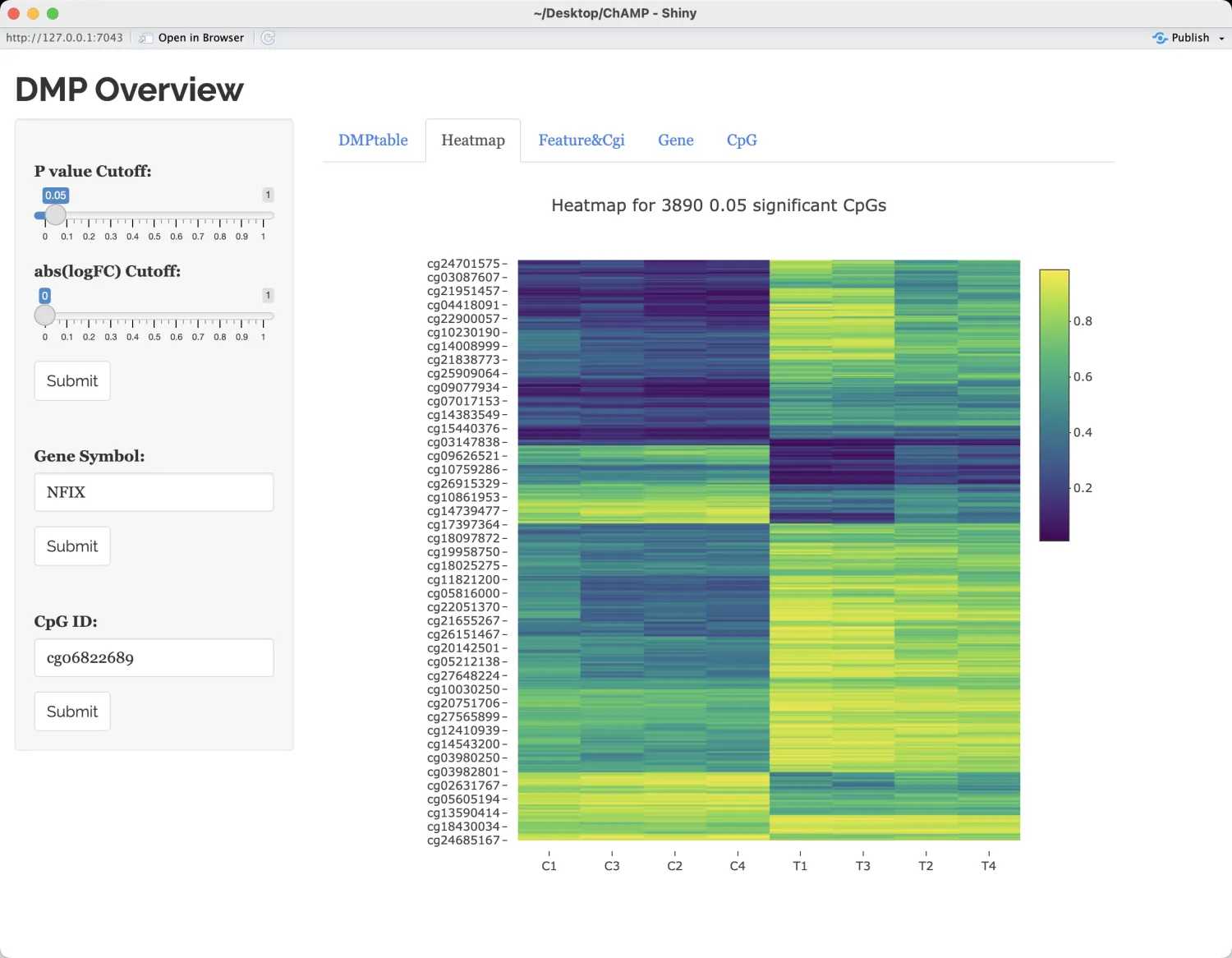
显示每种CpG特征(例如TSS200、TSS1500、body、opensea、shore等)以及CpG岛(CGI)总结信息在高甲基化(hyperCpGs)和低甲基化(hypoCpGs)中的比例分布。
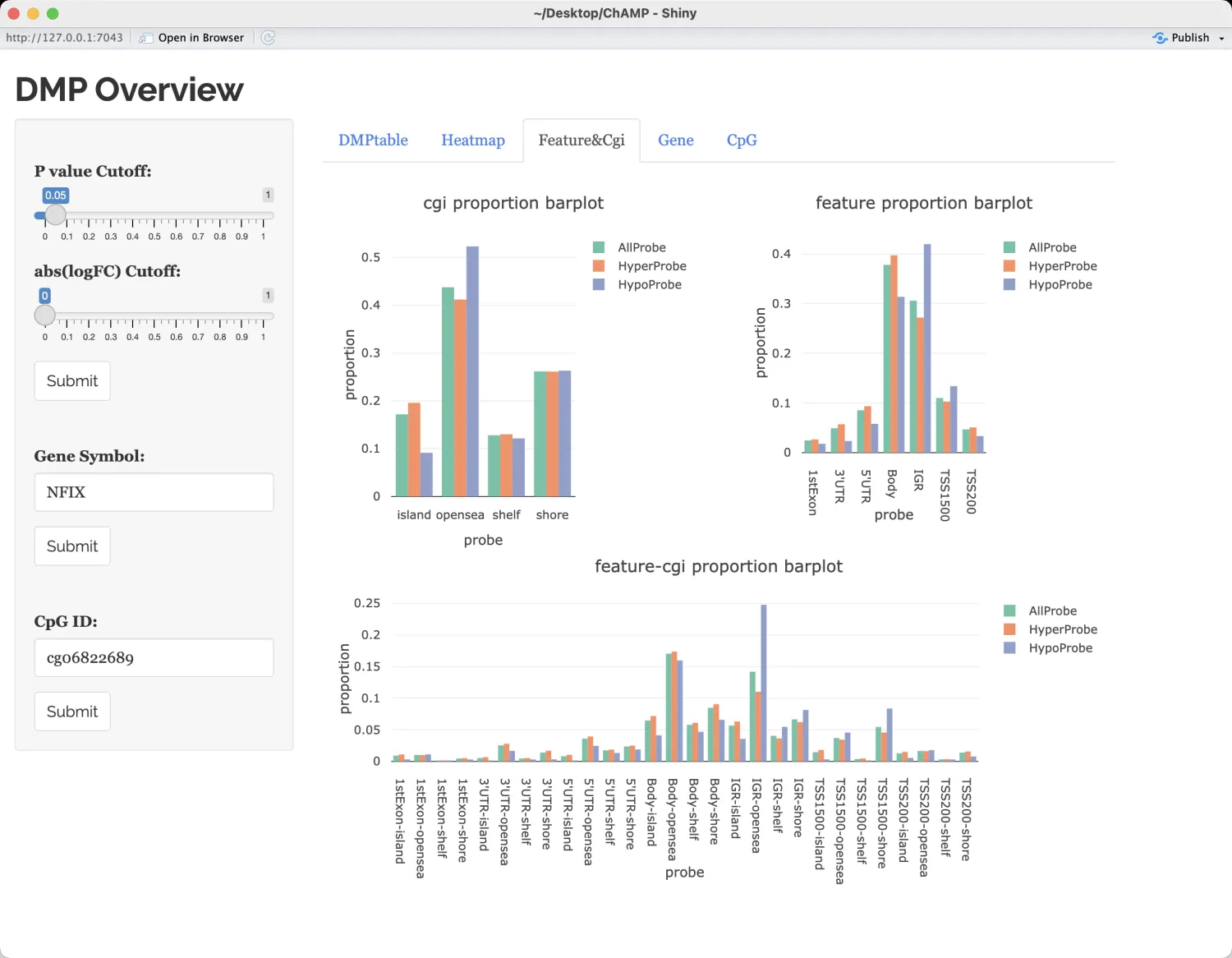
检测默认基因NFIX,相比于对照样本,该基因在肿瘤组织中具有更高的甲基化(挺有意思的,通常肿瘤中的甲基化情况比正常组织中要低)。
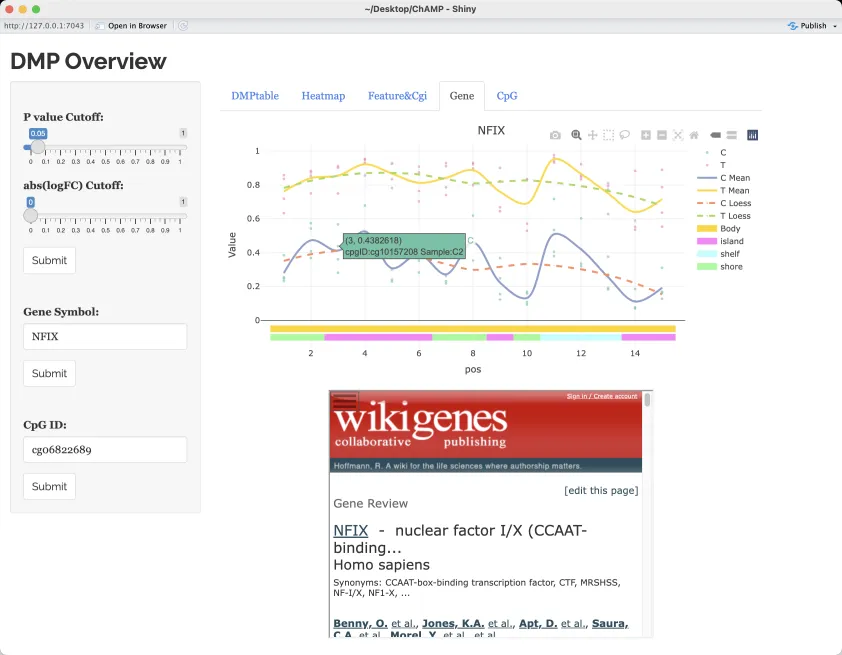
差异甲基化区域
差异甲基化区域(Differentially Methylated Regions, DMRs)是指基因组中在两组样本之间表现出DNA甲基化水平定量变化的扩展片段。ChAMP提供的champ.DMR函数用于计算和返回包含探针数据的差异甲基化区域(DMRs)数据框,并附带相应的P值。
在ChAMP中实现了三种DMR算法:Bumphunter、ProbeLasso和DMRcate。
- Bumphunter算法首先将所有探针分组为较小的簇(或区域),然后使用随机置换方法来评估候选DMRs。该方法非常用户友好,并且不依赖于之前函数的输出。置换步骤可能会消耗较多的计算资源,但用户可以分配更多的处理核心来加速计算。Bumphunter算法的结果是一个包含所有检测到的DMRs的数据框,其中包括其长度、簇信息以及注释的CpG位点数目。
- ProbeLasso方法的最终数据框是从limma输出的探针关联统计结果中提炼而来。之前,用户需要将champ.DMP()的结果输入到champ.DMR()函数中以运行ProbeLasso,但现在函数已升级,不再需要依赖champ.DMP()的输出。关于ProbeLasso方法的原理与机制,用户可以参考其原始论文。
- 最近整合到ChAMP中的新方法是DMRcate,这是一种基于数据驱动的方法,对所有注释均保持不可知态度,但对空间注释(如染色体坐标)敏感。DMRcate的核心是利用limma生成的单个CpG位点的差异甲基化(DM)稳健估计值。DMRcate将每个450K探针计算出的调节t统计量的平方输入到DMR检测函数中。然后,DMRcate使用高斯核在给定窗口内平滑这一指标,同时通过不同CpG密度引起的代表性减少和不规则间距来推导平滑估计值的期望值(即没有实验效应时的平滑值)。
注意事项:在当前版本的ChAMP中,在champ.DMR中增加了一些严格的检查步骤。champ.DMR现仅接受具有完全两类表型的分类变量。如果分类变量包含多类表型(如“肿瘤”/“转移”/“对照”),请手动选择其中任意两类,并输入相应的beta矩阵和表型信息以进行分析。
myDMR <- champ.DMR(beta=myNorm,pheno=myLoad$pd$Sample_Group,method="Bumphunter")
DMR.GUI(DMR=myDMR)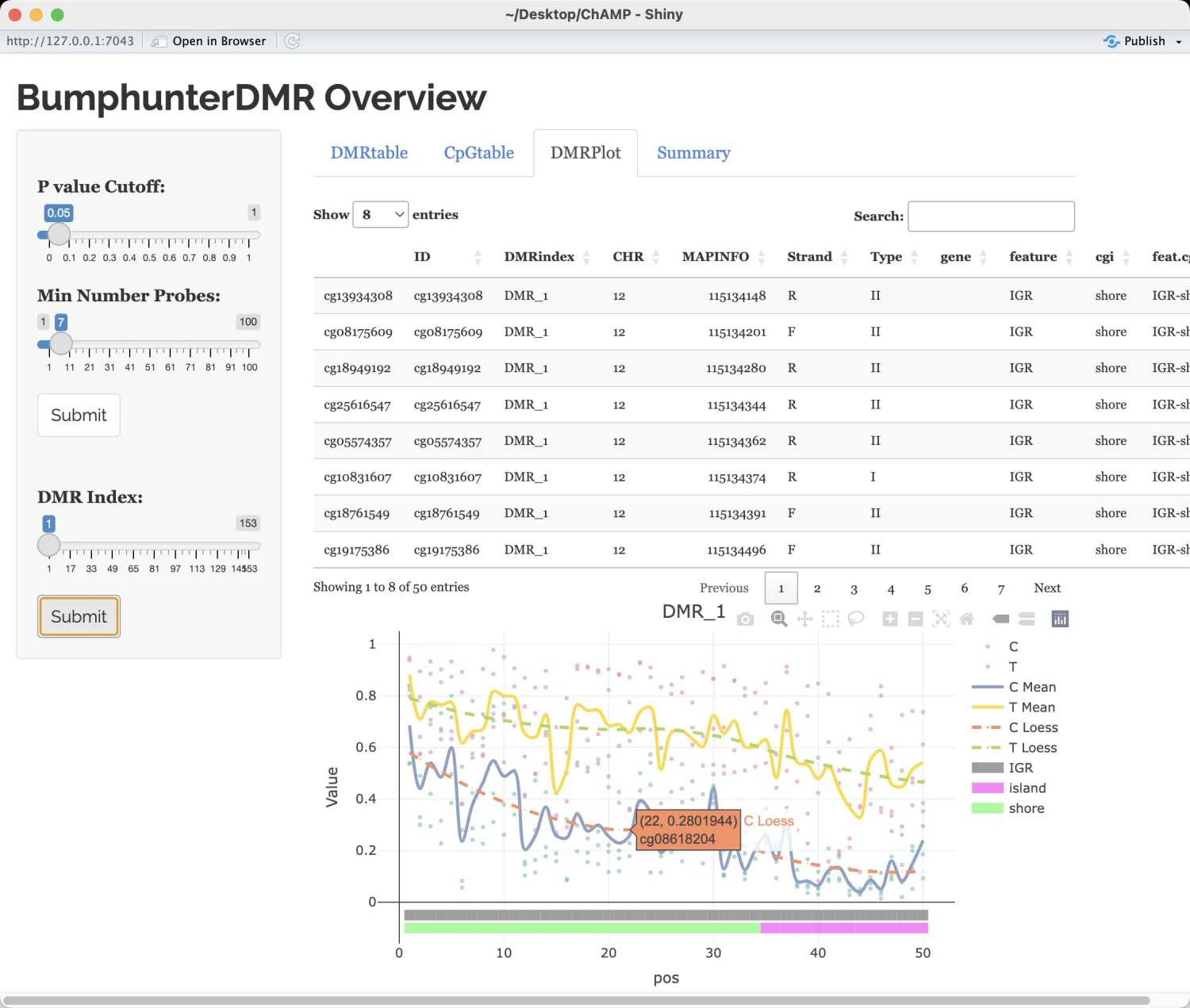
差异甲基化Blocks
差异甲基化块(Differentially Methylated Blocks, DMBs)的研究近年来逐渐受到关注。开发者提供了一个函数用于推断DMBs。在块检测函数champ.Bloc中,首先基于基因组上的位置对全基因组进行小簇(区域)的划分。然后,对于每个簇(区域),计算其平均值和平均位置,从而可以将每个区域简化为单个单位。在寻找DMB时,只有来源于opensea的单个单位会被用于聚类分析。接着应用Bumphunter算法在这些经过简化的区域(单位)上检测差异甲基化区域(DMRs)。
myBlock <- champ.Block(beta=myNorm,pheno=myLoad$pd$Sample_Group,arraytype="450K")
Block.GUI(Block=myBlock,beta=myNorm,pheno=myLoad$pd$Sample_Group,runDMP=TRUE,
compare.group=NULL,arraytype="450K")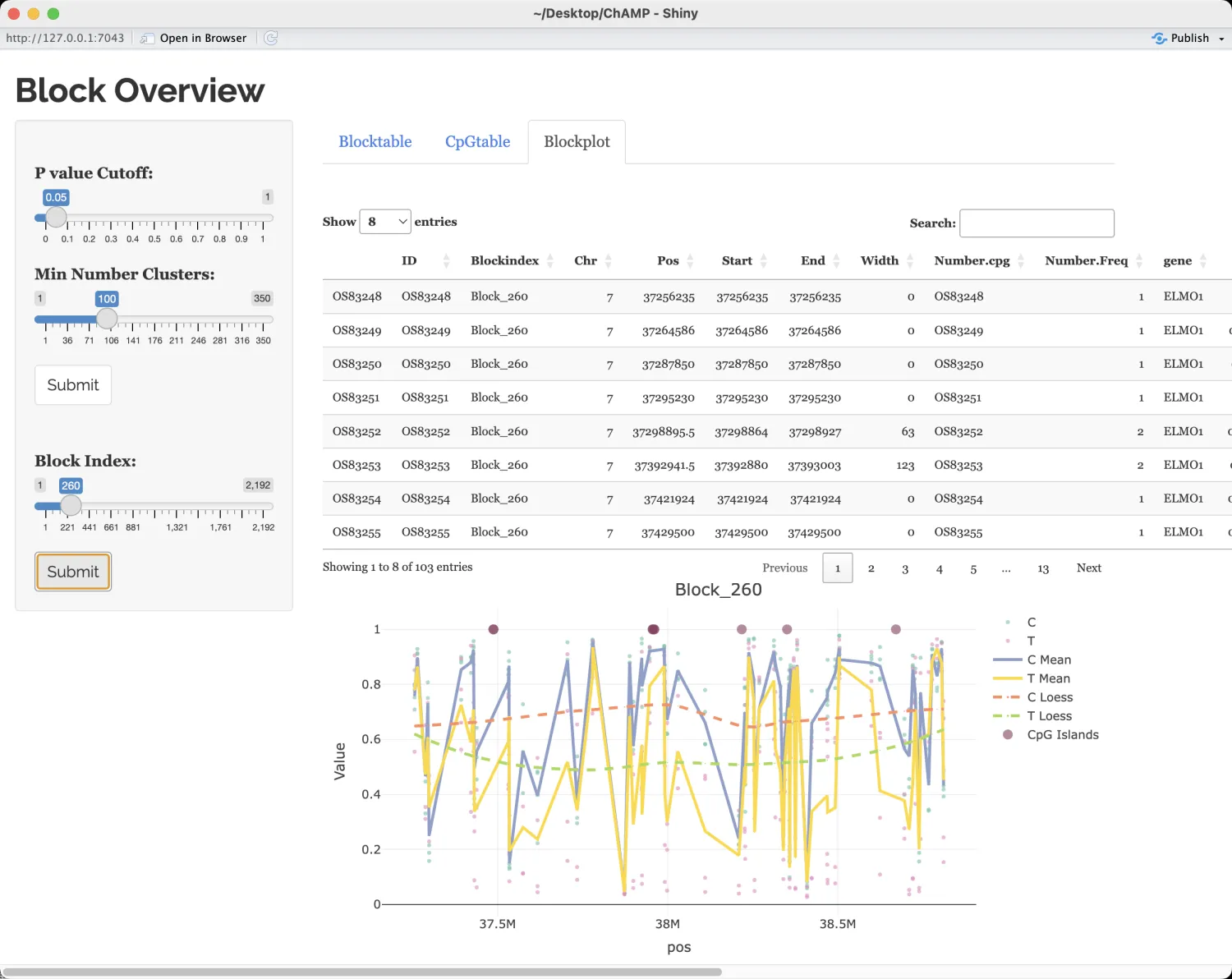
总之,差异甲基化探针(DMPs)、差异甲基化区域(DMRs)和差异甲基化块(DMBs)是DNA甲基化分析的三个层次。DMPs聚焦于单个位点的甲基化水平差异,适合精准识别关键CpG位点;DMRs通过分析相邻CpG位点的联合变化,揭示基因调控区域的甲基化特征;DMBs则覆盖更大范围的基因组区域,强调大规模甲基化变化的全局性特征,常用于复杂疾病的表观遗传研究。这三者从单个位点到大范围区域,粒度逐渐增大,分析目的从局部精准到全局概览,各有侧重且互为补充。
富集分析
champ.GSEA 会自动提取基因信息,将CpG位点信息转换为基因信息,然后对每个列表进行GSEA分析。在CpG到基因的映射过程中,如果某基因对应多个CpG位点,该基因只会被计算一次,以避免重复计算。GSEA分析有三种实现方式:
- Fisher Exact Test: 在之前的ChAMP版本中,基于从MSigDB下载的通路信息,使用Fisher精确检验计算每条通路的富集状态。基因富集分析完成后, champ.GSEA() 会自动返回P值小于设定阈值的通路。
- gometh方法: Geeleher等研究指出,由于基因中包含的CpG位点数目存在差异,例如某基因含有50个CpG位点但仅一个位点显著甲基化,另一基因仅有2个CpG位点但两个都显著甲基化,这两种情况不应被同等对待。为解决此问题,可以使用 missMethyl 包中的 gometh 函数,该函数根据基因中CpG位点的数量,而非基因长度,来校正这一偏倚。其核心思路是在所有基因中拟合一个CpG数量分布曲线,并利用概率加权函数修正GO分析的P值。
- 经验贝叶斯方法(ebayes): 在最近版本的ChAMP中,新增了基于经验贝叶斯的GSEA方法,可通过 method="ebayes" 参数调用。此方法无需依赖DMP或DMR结果,而是通过全局检验直接识别显著基因,并校正基因中CpG数量的不均衡。此外,该方法考虑了每个CpG显著性的程度,因此用户可以通过此方法检测边缘显著的基因以用于GSEA分析。
注意: 如果希望校正基因中CpG数量的不均衡偏倚,同时考虑CpG显著性水平,可以将 method 参数设置为 "ebayes" 使用经验贝叶斯方法。否则,可以选择 "gometh" 方法或 "fisher" 方法进行GSEA分析。
1)基因集富集分析
#基因集富集分析
myGSEA <- champ.GSEA(beta=myNorm,DMP=myDMP[[1]], DMR=myDMR, arraytype="450K",adjPval=0.05, method="fisher")
head(myGSEA$DMP$Gene_List)
# [1] "BENPORATH_ES_WITH_H3K27ME3" "TRANSC_FACT" "CAGGTG_V$E12_Q6"
# [4] "CTTTGT_V$LEF1_Q2" "BENPORATH_SUZ12_TARGETS" "BENPORATH_EED_TARGETS"
head(myGSEA$DMR$Gene_List)
# [1] "BENPORATH_EED_TARGETS" "TCCAGAG,MIR-518C" "ATATGCA,MIR-448"
# [4] "BENPORATH_ES_WITH_H3K27ME3" "GGGACCA,MIR-133A,MIR-133B" "chr5q31" 2)经验贝叶斯GSEA方法
myebayGSEA <- champ.ebGSEA(beta=myNorm,pheno=myLoad$pd$Sample_Group,arraytype="450K")
myebayGSEA$EnrichGene$GARGALOVIC_RESPONSE_TO_OXIDIZED_PHOSPHOLIPIDS_BLACK_DN
#[1] "MTSS1" "TARS2" "OSGEPL1" "ZNF22" "ZEB2" "SLC30A1" "ELAC1" "SMAD3" "ABHD10" "PIK3C2B"
# CNV分析CNV分析
ChAMP工具包提供了champ.CNA函数。此函数使用HumanMethylation450或HumanMethylationEPIC数据检测拷贝数变化。通过利用每个探针的强度值来计算拷贝数并确定是否存在拷贝数变异。拷贝数的计算基于CopyNumber包实现。
基本上,有两种方法可以用于CNA分析:第一种方法是比较病例样本与对照样本之间的拷贝数变化;第二种方法是比较每个样本的拷贝数状态与所有样本的平均拷贝数状态。对于第一种方法,用户可以在pheno参数中指定样本组,或者直接使用ChAMP内置的血液对照样本作为对照组进行拷贝数变异计算。若要使用内置对照组,只需将 control=TRUE 和 controlGroup="champCtls" 赋值即可。对于第二种方法,则可设置 control=FALSE。
champ.CNA会生成两种类型的图:单个样本分析(通过sampleCNA=TRUE参数控制)和每组样本分析(通过 groupFreqPlots=TRUE 参数控制)。与champ.QC函数类似,该函数提供了两个参数用于图形绘制:Rplot 参数用于控制是否在R会话中绘制图形,而 PDFplot 参数用于控制是否将PDF格式的图形保存到 resultsDir。默认情况下,仅 PDFplot 参数为TRUE。
需要注意的是,与旧版本的ChAMP不同,champ.CNA 不会自动对强度数据进行批次校正。如果需要对强度数据进行批次效应校正,请使用 champ.runCombat 函数。对于强度数据的使用方式,与Beta值或M矩阵完全相同,仅需将Beta值替换为 myLoad$intensity,并将 logitTrans=FALSE。
myCNA <- champ.CNA(intensity=myLoad$intensity,pheno=myLoad$pd$Sample_Group
,Rplot=T)
myCNA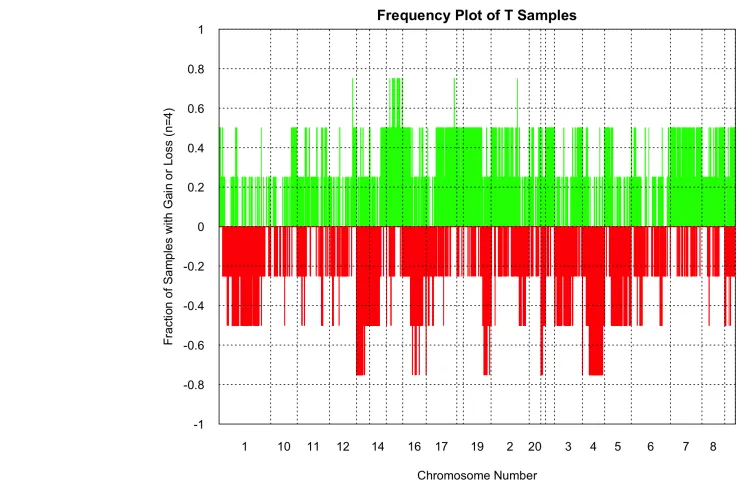
参考资料:
- ChAMP github:https://github.com/YuanTian1991/ChAMP
- ChAMP pipline introduction:https://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html
- Bisulfite sequencing (BS-seq) is the current "gold-standard" technology for high-resolution profiling of DNA methylation.
- Reduced representation bisulfite sequencing (RRBS) is an efficient form of BS-seq that targets CpG-rich DNA regions in order to save sequencing costs.
- 生信技能树:https://mp.weixin.qq.com/s/YRLeI-rvMcHQCfLVqkH_ZA https://mp.weixin.qq.com/s/YCoc4eirQ4Dxe2kYPd6oGw https://mp.weixin.qq.com/s/VG_MSD8_9HXG1YcW1_xShA https://mp.weixin.qq.com/s/3Xiln2CI2ZLTmAVSKWGmsw https://mp.weixin.qq.com/s/UbN8PONb07HD4hEhJ-GzXw https://mp.weixin.qq.com/s/nMDf4hHhhrX65VqD5WpF0g https://mp.weixin.qq.com/s/-E50Jvzo8aNqVgvEB0nVGA
- Epigenetics表观遗传学:https://mp.weixin.qq.com/s/nsIyDnaePCEhwJxW7-Zm1A
- 医学和生信笔记:https://mp.weixin.qq.com/s/O_W-P_HpziXtNMZXZm8b4w https://mp.weixin.qq.com/s/1xpT1E4BaWG-ulrCzylwrA
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号