从热力学视角重新审视药物分子的结合之道

从热力学视角重新审视药物分子的结合之道

DrugIntel
发布于 2026-05-20 12:47:56
发布于 2026-05-20 12:47:56

文献信息
- • 标题:Applying thermodynamic profiling in lead finding and optimization
- • 作者:Gerhard Klebe(德国马尔堡大学药物化学研究所)
- • 期刊:Nature Reviews Drug Discovery
- • 发表时间:2015 年 2 月
- • DOI:10.1038/nrd4486
导言:为什么结合亲和力不足以告诉你全部?
在经典的药物化学工作流程中,研究者通常以 IC₅₀、Kd 或 Ki 作为优化配体结合能力的核心指标。这些数值通过酶学抑制实验或结合竞争实验获得,操作简便、通量较高,是先导化合物筛选和结构-活性关系(SAR)研究的主力工具。
然而,这些数值所反映的,本质上只是吉布斯自由能(ΔG)的一个单一数字。而 ΔG 本身可以分解为两个物理意义截然不同、却相互竞争的热力学分量:
- • ΔH(焓变):反映配体与受体形成直接相互作用(氢键、盐桥、范德华接触)以及伴随的去溶剂化过程中能量的净变化。
- • TΔS(熵贡献):反映系统无序度的变化,涵盖结合时构象自由度的损失、水分子的捕获或释放、配体和蛋白质残余动态行为等多方面因素。
两个 ΔG 完全相同的配体,其 ΔH 和 TΔS 的分配可能截然不同——一个可能是强焓驱动、弱熵驱动,另一个恰恰相反。Klebe 在本文中系统论证了:这种"热力学特征"(thermodynamic signature / thermodynamic profile)对于理解结合机制、指导结构优化、预测抗耐药性,乃至判断苗头化合物的优化潜力,都具有不可忽视的实用价值。
一、热力学基础:从自由能到焓熵分解
1.1 结合常数与自由能
在化学平衡条件下,配体(L)与受体(P)的结合过程可用解离常数 Kd 描述:
在标准态、恒压条件下,Kd 与 ΔG 的关系为:
其中 R 为理想气体常数,T 为绝对温度。由此可见,Kd 每降低一个数量级(即亲和力提升 10 倍),对应约 −5.7 kJ/mol 的 ΔG 改善(25°C 时)。
药物化学优化通常需要将 Kd 从毫摩尔级(苗头片段)推进至纳摩尔甚至皮摩尔级(候选药物),对应 ΔG 从约 −15 kJ/mol 提升至 −60 kJ/mol 的范围——这正是可被化学合理地访问的热力学窗口。

1.2 IC₅₀ 与 Ki 的区别及其局限性
值得注意的是,实验中更常用的 IC₅₀ 并不等同于 Kd 或 Ki:
- • IC₅₀ 依赖于底物浓度和酶的浓度,是条件依赖性参数;
- • 通过 Cheng-Prusoff 方程可将 IC₅₀ 转换为 Ki,但该转换需要已知底物的 Km;
- • 不同实验室使用不同底物浓度时,IC₅₀ 数据无法直接横向比较。
因此,仅依赖 IC₅₀ 进行 SAR 研究存在系统性盲区,而 ITC 直接测量的热力学参数提供了更本质的结合信息。
1.3 配体效率与焓效率
为了在提升亲和力的同时控制分子量,配体效率(LE)被广泛用于指导优化:
其中 N_heavy 为非氢原子数。以 LE > 0.3 kcal/mol/atom 作为苗头化合物的筛选基准已是行业惯例。
Klebe 进一步引入了焓效率(enthalpic efficiency)的概念:
其逻辑是:在优化过程中,熵贡献(如骨架刚性化、疏水填充)可以在晚期相对可预期地引入;而焓贡献的质量(每个原子贡献的焓值)更难通过化学修饰灵活调控。因此,优先选择焓效率高的苗头,为后续熵优化留出空间,是一种更稳健的策略。
二、ITC:测量热力学数据的金标准
2.1 工作原理
等温滴定量热法(Isothermal Titration Calorimetry, ITC)是目前唯一能在一次实验中同时、直接测定 ΔG 和 ΔH 的技术,无需标记或固定化。
实验中,已知浓度的配体被逐步滴入含有蛋白质的样品池,量热计实时检测每次注射后溶液温度的微小变化(通常为 μcal 级)。通过对滴定曲线的非线性拟合,可同时获得:
- • ΔH:直接由热信号测量;
- • Ka(或 Kd):由滴定曲线的 S 形特征确定;
- • ΔG:由 Ka 计算;
- • TΔS:由 ΔG − ΔH 计算。

2.2 核心缺陷:不可避免的焓-熵数学耦合
ITC 最重要的内在局限,往往被研究者忽视:
TΔS 不是独立测量的,而是通过 ΔG − ΔH 的数值差计算得到的。
这意味着:
- • 测量 ΔH 时的任何系统误差,都会被无差别地反映在 TΔS 中;
- • 即使没有任何物理意义的焓-熵补偿,纯粹的测量误差也会制造出数学上的焓-熵补偿假象。
这种"人为的焓-熵补偿"(artifactual enthalpy-entropy compensation)是解读 ITC 数据时最常见的陷阱之一。
2.3 质子化效应的校正:最易忽视的系统误差
当配体或蛋白质功能基团在结合过程中发生质子化状态变化时,缓冲液的电离热(heat of ionization)会叠加在结合热信号之上,造成 ΔH 的失真。
校正方法:在不同 heat of ionization 的缓冲液(如磷酸盐、Tris、HEPES 等)中分别进行 ITC 实验。若测得的 ΔH 随缓冲液变化,则说明存在质子交换;通过线性外推可分离出"纯"的结合焓。
关键结论:
- • 含氮功能基团(胺、咪唑等)的 heat of ionization 较大,校正尤为重要;
- • 含氧功能基团(羟基、羧基等)的 heat of ionization 较小,影响相对有限;
- • 若实验系列中质子化位点与化学修饰位点距离较远,可认为质子化贡献在系列内相对恒定,此时比较同系物间的相对差值仍具统计意义,即使未做绝对校正。
2.4 片段热力学的挑战
近年来基于片段的药物发现(FBDD)兴起,理想状态下应在苗头片段阶段就获取热力学信息以指导后续优化方向。然而,这在实践中面临严峻挑战:
- • 片段(MW 150–250 Da)的结合亲和力通常在毫摩尔级,产生的热信号极弱,难以形成 S 形滴定曲线;
- • 高浓度蛋白和片段的要求受限于溶解度和蛋白稳定性;
- • 非 S 形曲线须假设 1:1 化学计量比,而这对蛋白-片段体系往往不成立;
- • 片段结合位点周围大量水分子的重排,对热力学信号的贡献不可忽视,且难以独立量化。
目前,位移滴定协议(displacement ITC)是解决高 Kd 片段测量的较可行方案,但仍需高亲和力参比配体及严格的实验设计。
三、焓-熵补偿:药物优化中几乎无法回避的困境
3.1 物理本质
Olsson 等人汇编了来自药物化学和生物来源的大量 ITC 数据,绘制于 ΔH 对 −TΔS 的二维图中,揭示了一个普遍规律:
- • 主对角线方向:ΔG 的散布范围(约 −15 至 −60 kJ/mol),对应从苗头到候选药物的优化区间;
- • 垂直于主对角线方向:ΔH 和 −TΔS 的相互抵消(scatter),范围远大于 ΔG 的散布——这正是焓-熵补偿的直观体现。
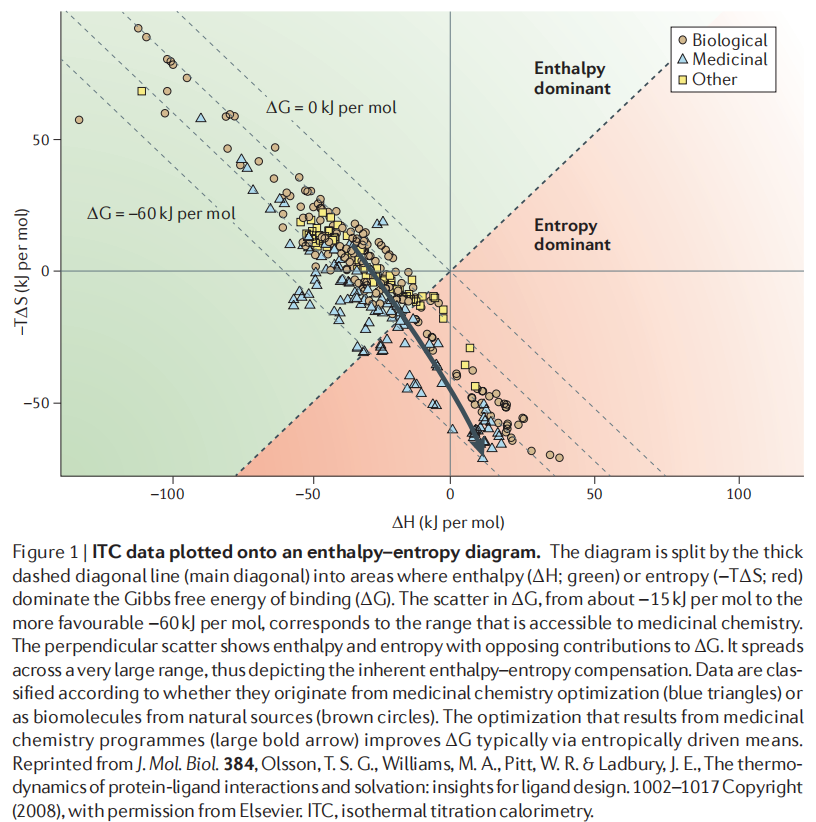
值得注意的是,来自药物化学优化的配体(蓝色三角形)相比生物来源分子(棕色圆圈),主要沿着熵优化方向(向红色区域)移动——也就是说,传统的药物化学优化往往在无意识地以熵驱动提升亲和力,而非焓驱动。
3.2 分子机制
焓-熵补偿的分子机制并非某个单一规律,而是多种物理效应的叠加:
优化操作 | 焓效应 | 熵效应 | 净效应解释 |
|---|---|---|---|
形成强氢键(深埋口袋) | ΔH ↓(有利) | −TΔS ↑(不利) | 固定配体降低残余移动性 |
引入带电基团(盐桥) | ΔH ↓↓(强有利) | −TΔS ↑↑(强不利) | 去溶剂化代价大,结合后自由度更受限 |
疏水基团填充口袋(驱替有序水) | ΔH 轻微变化 | −TΔS ↓(有利) | 水分子释放至体相增加系统熵 |
刚性化骨架 | ΔH 变化小 | −TΔS ↓(显著有利) | 减少结合时损失的构象自由度 |
捕获水分子形成水媒氢键 | ΔH ↓(有利) | −TΔS ↑(不利) | 水分子被固定带来熵代价 |
核心洞见:焓-熵补偿并非物理定律,但在实践中几乎不可避免,因为能强烈锚定配体的相互作用(好的焓贡献)必然同时限制其残余移动性(坏的熵贡献)。
3.3 真实数据:一个揭示性的对照实验
Klebe 课题组以凝血酶抑制剂系列为例,比较了两组同系物:
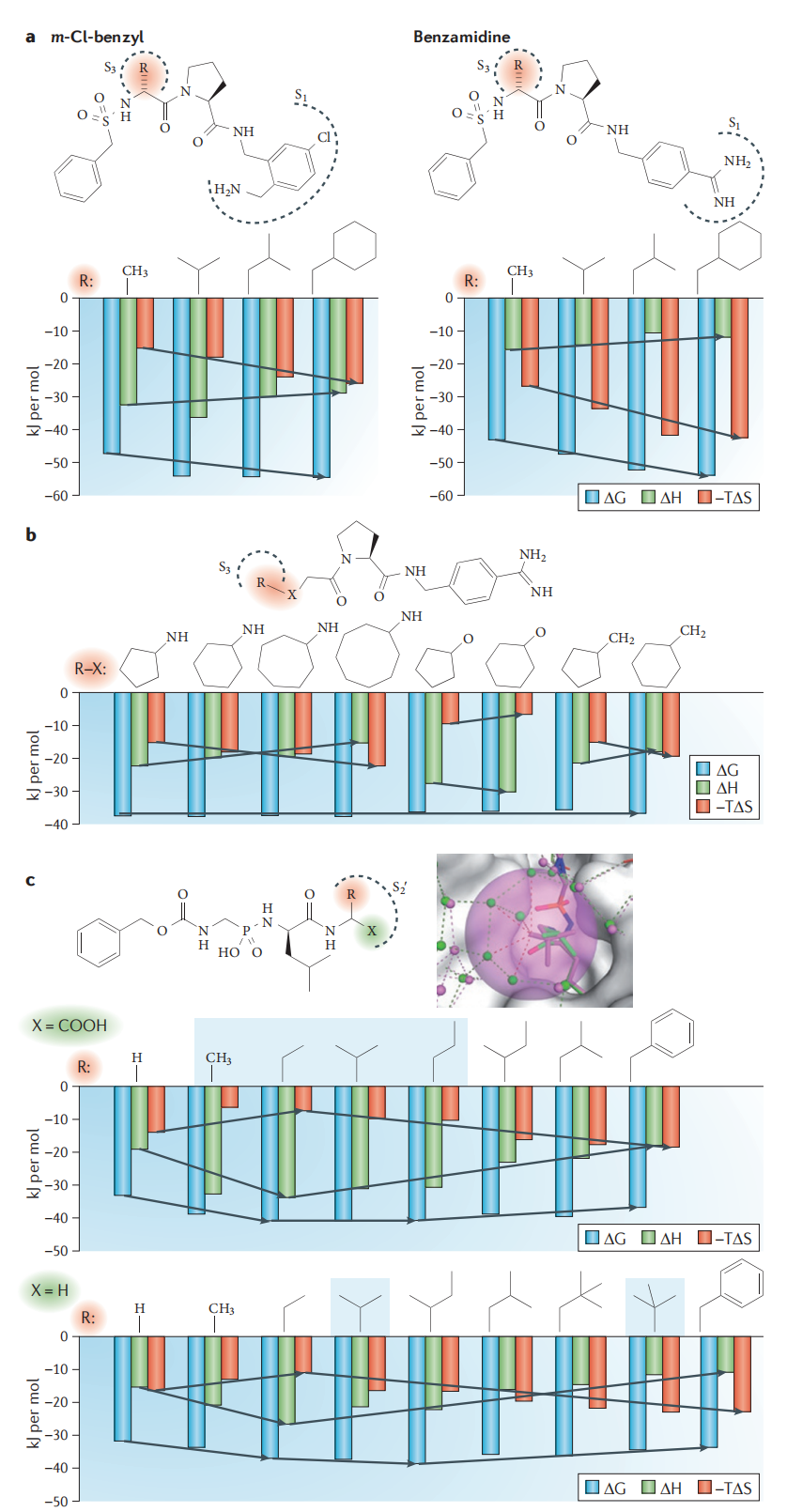

P2 位置刚性化对比:
- • 脯氨酸衍生物(P2 位刚性)vs. 甘氨酸衍生物(P2 位柔性)
- • 两者具有相同的结合几何构型
- • 脯氨酸衍生物相比甘氨酸:−TΔΔS = −18.4 kJ/mol(熵优势显著),ΔΔG = −23.6 kJ/mol
这一实验清晰表明:构象预组织(pre-organization)对亲和力的贡献主要来自熵,而非焓,且幅度可观。
四、氢键的热力学贡献:绝非简单叠加
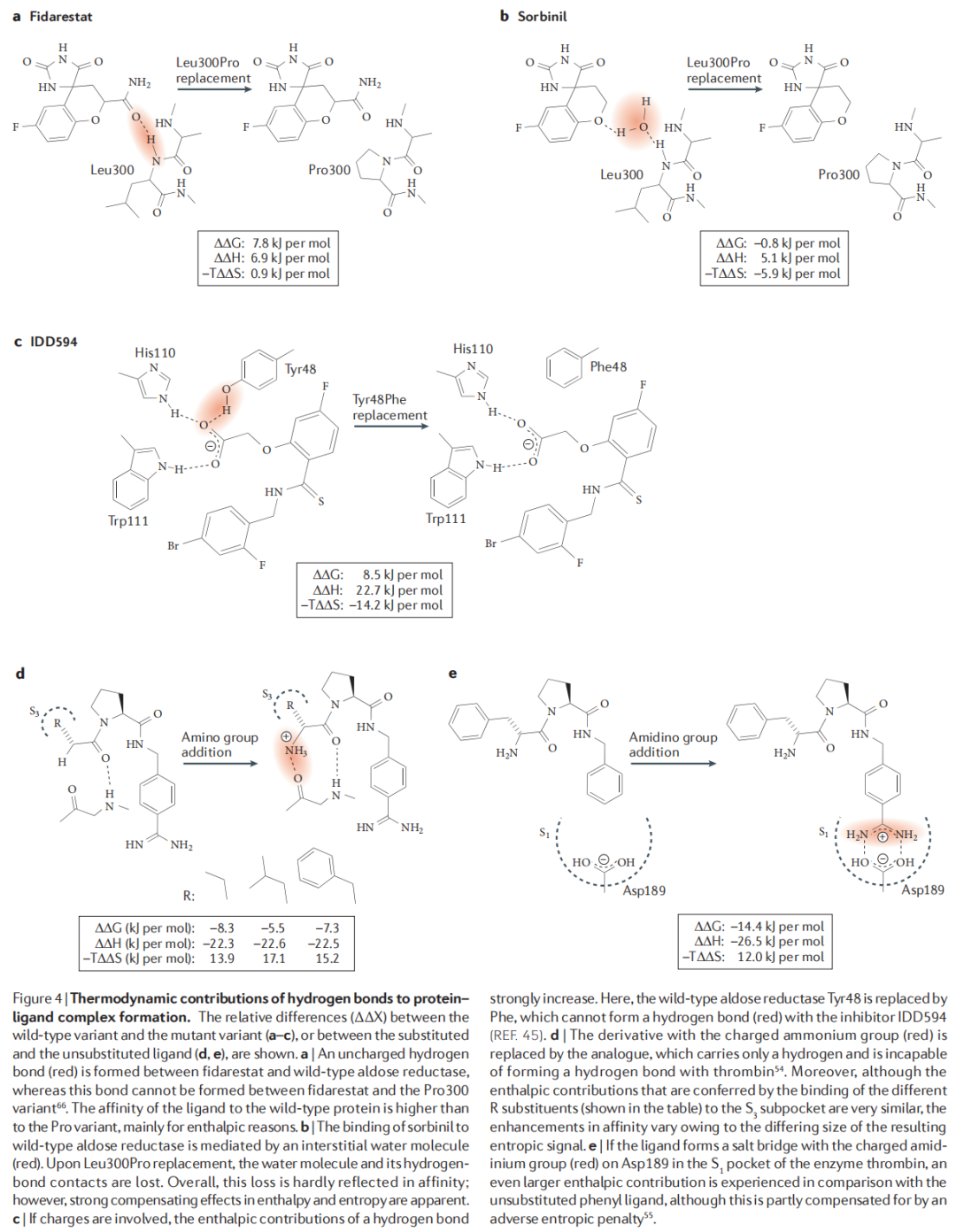
4.1 不带电氢键(中性氢键)
以 aldose reductase 为模型体系,通过 Leu300→Pro300 突变去除配体 fidarestat 与酶之间的一个主链 NH 氢键:
,,
结论:中性氢键主要为焓贡献,熵代价极小。
4.2 带电辅助氢键(charge-assisted H-bond)
同一口袋中,Tyr48→Phe48 突变去除配体 IDD594 与酶之间的带电辅助氢键:
,,
结论:带电氢键的焓贡献远大于中性氢键(约 3 倍),但伴随显著的熵代价——二者在 ΔG 层面相互抵消,总体 ΔG 增量与中性氢键相近。
4.3 盐桥
在凝血酶 S1 口袋引入苯脒基(amidinium)与 Asp189 形成盐桥:
,,
4.4 重要推论
这一系列实验揭示了一个在药物化学中常被误解的规律:
带电相互作用的焓贡献更大,但熵代价也更大,净 ΔG 改善并不总是优于中性氢键。
更关键的是,在凝血酶抑制剂系列研究中,带电辅助氢键的焓贡献在不同 R 基团取代的情况下几乎恒定(约 −22 kJ/mol),但总体亲和力的差异却因 R 基团带来的不同熵信号而产生显著变化。这说明:
- • 氢键的焓贡献受局部电荷环境控制,相对固定;
- • 氢键对整体亲和力的实际贡献,高度依赖于同一复合物中其他相互作用对熵的影响;
- • 功能基团的贡献不具有严格的加合性(non-additivity)。
五、疏水效应的热力学实质:并非总是熵驱动
5.1 经典疏水效应
经典疏水效应描述的是:非极性基团填充疏水口袋时,将原本有序排列的水分子驱替至体相,增加溶剂熵,从而以熵驱动的方式提升亲和力。
实验验证:以两系列凝血酶抑制剂为研究对象,P3 位疏水取代基从甲基逐步扩展至环己基甲基。随着亲脂性增加,亲和力主要以熵驱动方式提升,符合经典疏水效应的预期——前提是被驱替的水分子原本处于有序排列状态。
5.2 非经典疏水效应:焓驱动的疏水结合
然而,文章列举了多个焓驱动的疏水结合案例(来自 thermolysin 和碳酸酐酶体系),挑战了"疏水结合=熵驱动"的简单化认知:
- • 当被驱替的水分子原本处于非最优氢键网络("unhappy waters",即氢键形成不完整)中时,其释放对焓有正贡献;
- • 当填充的疏水口袋原本水化不足(suboptimally hydrated)时,配体结合同样可以表现出焓驱动特征。
这一发现的实践意义在于:WaterMap 等计算工具对水分子性质的预测(是否为"unhappy water")将直接影响对疏水结合热力学特征的预判,从而指导优化策略的选择。
六、氢键与疏水接触的协同效应
6.1 非加合性的实验证明
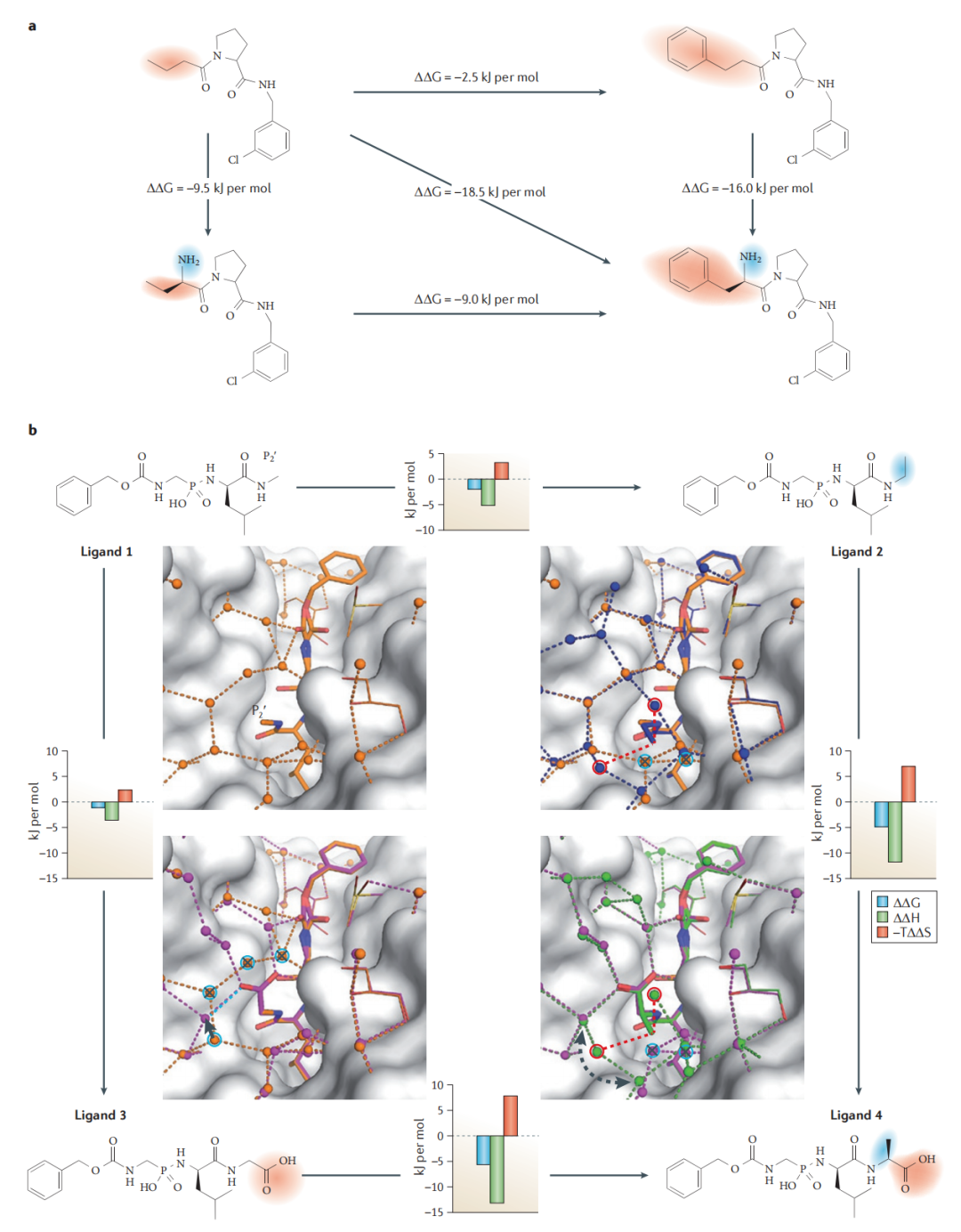
Klebe 课题组对凝血酶抑制剂系列进行了精心设计的 2×2 矩阵实验:以有序/无序地引入亲脂侧链和氨基基团(可形成带电氢键),逐步比较四种组合的热力学差异。
关键发现:
- • 先引入氢键再引入亲脂基团,与先引入亲脂基团再引入氢键,两种路径的逐步 ΔΔG 增量不同——即功能基团的贡献不满足加合性;
- • 氢键存在时,中等大小疏水取代基的亲和力贡献显著增大(表面积增量从 −78 J/mol/Ų 提升至 −128 J/mol/Ų)。
6.2 动态性质解释协同效应
分子动力学模拟揭示了这一协同效应的分子机制:
- • 当氢键缺失且疏水基团较小时,P3 取代基在 S3 口袋中具有较大的残余移动性,与蛋白质的疏水接触效率低下(时间平均接触面积减小);
- • 氢键的引入限制了取代基的残余移动性,使疏水接触更为稳定高效;
- • 当疏水基团足够大时,它本身就能通过空间填充限制自身移动,此时氢键的协同增益减小。
这一发现提示:孤立地考察单个相互作用的贡献是不充分的;复合物的动态行为(residual mobility)是理解热力学特征的核心变量。
七、水分子:结合界面的隐形设计师
7.1 结合位点内的水分子
水分子在配体结合位点的行为是决定热力学特征(特别是焓/熵分配)最重要、也最难预测的因素之一。
Aldose reductase 模型系统中,两个结构高度相似的等效能抑制剂呈现截然不同的热力学特征:
- • Ligand 1:捕获一个中介水分子形成水媒氢键(water-mediated H-bond),呈焓主导;
- • Ligand 2:不涉及中介水分子,呈熵主导;
- • 二者 ΔG 几乎相同,但 ΔH 和 TΔS 差异悬殊——仅凭亲和力数据完全无法区分。
Sorbinil 与野生型/Leu300Pro 突变型 aldose reductase 结合的对比研究进一步确认:
- • 野生型:水媒氢键形成(焓有利 −5.1 kJ/mol),但捕获水分子付出熵代价;
- • Pro300 突变体:中介水分子消失(去除了捕获成本),熵有利,但失去了焓增益;
- • 两者 ΔG 几乎相同,但焓-熵特征完全相反。
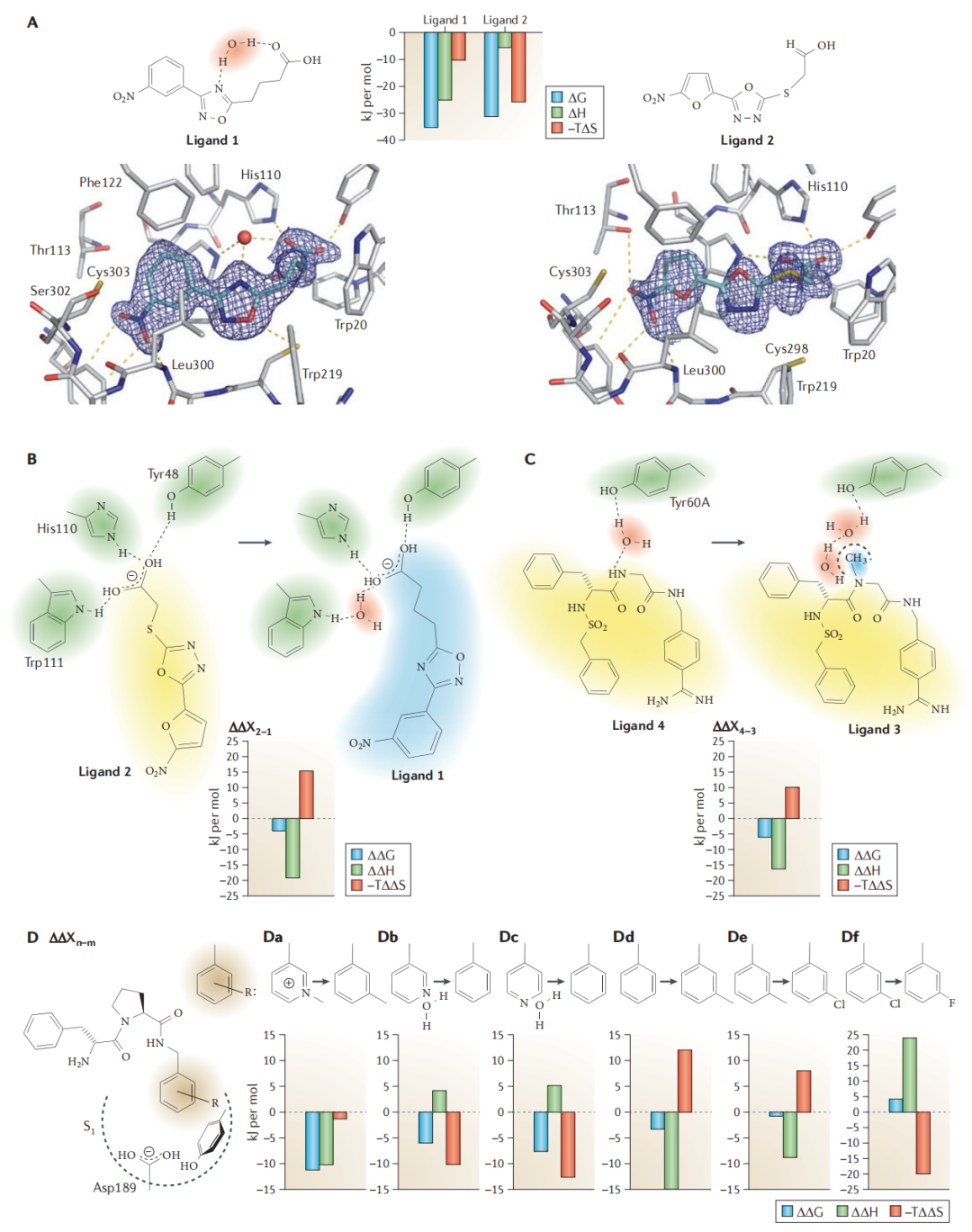

7.2 复合物表面的水网络
更深刻的发现来自 thermolysin 系列研究。该体系的配体系列在 P2ʹ 位置引入不同大小的疏水取代基,所有配体采用相同结合模式,但复合物表面的水网络结构随取代基大小系统变化。
关键实验结果(Biela 等人,2012–2014):
取代基 | 水网络完整性 | ΔG 趋势 | 主导特征 |
|---|---|---|---|
甲基 | 引入,优化网络 | 改善约 −2 kJ/mol | 轻微焓有利 |
异丙基 | 近乎完美网络 | 最优 | 焓主导 |
叔丁基 | 网络破坏,水分子被驱替 | 劣于异丙基 | 熵主导 |
苄基/异丁基 | 大空间位阻,大量驱水 | 亲和力下降 | 熵主导但总体不利 |
最反直觉的发现:仅仅增加一个甲基(从异丙基到叔丁基),热力学特征就从焓主导逆转为熵主导,而这一变化的根源在于表面水网络的精细结构被扰动,而非直接的配体-蛋白接触改变。
这彻底颠覆了"疏水效应由埋藏面积决定"的简化模型:表面水网络的质量(而非疏水面积的大小)才是决定热力学特征的关键变量。
7.3 非加合性再探:水网络的中介作用
在羧酸取代系列(thermolysin 研究)中,以不同顺序引入甲基和羧基,两条优化路径表现出不同的 ΔΔG 增量,但最终产物(同时具有甲基和羧基的配体)在两条路径中完全相同:
- • 路径 A(先甲基后羧基):两步增量均显著;
- • 路径 B(先羧基后甲基):中间体(仅有羧基)亲和力反而劣于母体(无羧基),因为引入带电基团破坏了已有的水网络,产生净的不利效果;随后的甲基化"修复"了水网络,才带来预期的亲和力增益。
这一发现具有重要的方法论意义:在不了解水网络结构的情况下,热力学数据看似矛盾的非加合性,可能恰恰揭示了水分子重排的关键信息。

八、在药物发现中的实际应用与局限性
8.1 热力学数据的用途
根据文章的系统总结,热力学分析在药物发现中具有以下实际价值:
(1)区分机制相同、亲和力相似的化合物
当 SAR 曲线趋于平坦(flat SAR),仅凭亲和力数据无法区分结合行为时,热力学数据可以揭示不同化合物在结合口袋中的行为差异——包括不同的结合构象、蛋白质构象变化或溶剂化模式的改变。
(2)指导苗头化合物的优化方向
从理论最优策略出发:应优先选择焓驱动的苗头(即高焓效率的化合物),因为:
- • 纳摩尔级以下亲和力的提升几乎不可避免地依赖熵贡献(骨架刚性化、疏水填充);
- • 以焓为起点,可以在晚期系统地引入熵贡献,而不必担心已有焓基础的流失;
- • 反之,若苗头已经是纯熵驱动,后续再引入大量焓贡献的可能性较低(因焓-熵补偿的限制)。
(3)检测质子化变化,指导 pKa 设计
在不同缓冲液下进行 ITC 实验,若 ΔH 随缓冲液变化,可定量分析配体或蛋白质功能基团在结合时的质子化/去质子化行为,从而指导对配体酸碱性(pKa)的定向调控,以优化生物利用度。
(4)追踪同系列中的异常变化
当同系列中某个化合物的热力学特征与其他成员出现突变时,往往预示着结合模式的变化(如结合口袋区域不同、蛋白质构象变化)或叠加效应(如额外的质子化变化)——这类信号在仅关注亲和力时将被完全忽略。
8.2 关键局限性与误区
文章对热力学分析的局限性保持了罕见的诚实,这些局限性同样是实践者必须了解的:
(1)不能在没有结构信息的情况下解读绝对热力学特征
将一个配体分类为"焓主导"还是"熵主导",离开了高分辨率晶体结构(及溶剂化信息)几乎没有预测价值。水分子重排可以在不改变 ΔG 的情况下,使 ΔH 和 TΔS 各偏移 ±5–10 kJ/mol。
(2)不同化合物系列之间的比较高度危险
以"更焓驱动的苗头更有优化潜力"为唯一判据,在比较不同 P1 锚基的凝血酶抑制剂系列时会导致错误结论:两个系列的相对焓/熵特征,仅由 P1 基团的属性决定,而非由所附加的变量基团决定。
(3)片段阶段的热力学数据可靠性极低
如前文所述,片段结合的弱热信号、水网络的高度参与、化学计量假设的不可靠性,使片段阶段的 ITC 数据在没有严格实验设计的情况下几乎不可信。
(4)ITC 测量的是整体结合事件
ITC 观测到的热力学参数是整个结合过程(包括配体去溶剂化、蛋白质去溶剂化、构象变化、水分子重排等)的综合结果。将总 ΔH 和 TΔS 分解至单个化学相互作用,在实践中极为困难,甚至不可能。
九、对抗耐药性的热力学策略
文章在引言部分引出了热力学策略的一个重要应用方向:抗病毒/抗菌药物的耐药性规避。
病原体产生耐药性的分子机制包括:
- • 空间错配(steric mismatch):突变位点直接改变结合口袋形状;
- • 蛋白质动态变化(protein dynamics):突变改变结合口袋的柔性,影响配体构象适应;
- • 氢键损失:突变导致关键相互作用基团的丢失。
Freire 等人提出,焓优化的、结合模式具有足够柔性的配体,对靶标基因突变引起的结合位点变化具有更强的耐受性——因为这类配体能够通过微调自身结合模式来适应局部变化,而不会因某一特定相互作用的丧失导致亲和力完全崩塌。
HIV 逆转录酶抑制剂的临床数据印证了这一理论:dapivirine 和 etravirine 相比同类其他抑制剂表现出更低的耐药发展速率,而这与它们能够在结合位点内以多种模式结合(multiple binding modes)密切相关——这正是熵有利的高残余移动性结合所赋予的特性。
十、综合框架:热力学指导的优化路线图
综合全文的实验数据与理论分析,可以构建如下热力学指导的先导化合物优化框架:
阶段一:苗头发现(Hit Finding)
- • 工具:ITC(需充分校正质子化效应)+ 高分辨率晶体结构
- • 目标:识别焓效率(HE)高的苗头,建立热力学基线
- • 注意:片段阶段数据的可靠性需格外审慎评估;水分子对热力学特征的影响需通过晶体结构追踪
阶段二:早期优化(Early Optimization, mM → μM)
- • 策略:优先在深埋口袋中引入氢键(增加焓贡献)
- • 监控:追踪同系列相邻化合物间的 ΔΔH 和 ΔΔS;关注非加合性信号
- • 机制洞见:带电氢键焓贡献大但熵代价高;中性氢键净贡献更可预测
阶段三:中期优化(Mid-stage Optimization, μM → nM)
- • 策略:向疏水口袋引入亲脂基团;关注被驱替水分子的性质(有序/无序)
- • 工具:WaterMap 等计算工具辅助预测水分子性质;结合 MD 模拟理解残余移动性
- • 注意:避免破坏已建立的表面水网络,尤其在亲脂取代基从中等向大型过渡时
阶段四:后期精细化(Late-stage Optimization, nM → pM)
- • 策略:骨架刚性化(引入脯氨酸类刚性连接、形成大环、减少旋转键)
- • 驱动力:主要来自熵贡献(减少结合时的构象损失)
- • 监控:确保刚性化引入的同时不对已有的焓网络造成破坏
十一、方法论建议:如何正确使用 ITC 数据
根据全文的讨论,Klebe 归纳了若干重要的方法论原则,适用于工业界和学术界的实践者:
- 1. 始终在多种缓冲液体系中进行 ITC,以检测并校正质子化效应,避免虚假焓-熵补偿。
- 2. 优先比较同系列内的相对差值(ΔΔG、ΔΔH、ΔΔS),而非绝对数值,以最小化系统误差的影响。
- 3. 不能仅凭热力学数据做决策,必须结合高分辨率晶体结构(理想状态下还需溶剂化结构信息),才能将热力学信号分配给具体的结构特征。
- 4. 热力学特征异常是有用的信号,当某个化合物的 ΔH/TΔS 特征明显偏离系列趋势时,应视为潜在的结构/机制变化的预警,而非舍弃数据。
- 5. 不应将"更焓主导=更优"简单化,因为 P1 锚基等固定基团对热力学特征的贡献,可能完全掩盖可变取代基的贡献,导致跨系列比较失去意义。
- 6. 对计算预测(ΔG 模拟)保持审慎:即使计算方法忽略了水分子,也往往能给出合理的 ΔG 预测——正是因为焓-熵补偿使总自由能对水结构不敏感,但这并不意味着计算方法真正理解了结合机制或能正确预测结合模式。
十二、总结与展望
Klebe 的这篇综述具有多层次的学术价值:
对实验药物化学家:它提供了一套系统的框架,将 ITC 数据整合进 SAR 工作流程,并明确指出了应用边界——热力学数据不是万能的,但在特定情境下(同系列比较、质子化变化检测、水分子效应识别)提供了亲和力数据所无法给出的信息。
对计算化学家:它强调了水分子精细结构对热力学特征的决定性影响,为 WaterMap、FEP 等计算工具的验证和改进提供了丰富的实验参照。
对药物发现项目团队:它论证了在先导化合物选择阶段引入热力学表征的合理性,尤其是在多个亲和力相当的苗头化合物之间做抉择时,热力学特征可以作为评估"优化潜力"的辅助维度。
尚待解决的核心问题:
- • 如何系统地将热力学特征分解至单个相互作用?
- • 片段阶段的热力学表征是否存在可靠的实验解决方案?
- • 动态结合模式(配体在口袋内多模式存在)如何在热力学测量中被定量区分?
- • 人工智能/机器学习模型能否在不依赖完整晶体结构的情况下预测水网络对热力学特征的影响?
这篇文章问世十年后,随着低温电镜技术的进步、分子动力学模拟算力的提升,以及基于 AI 的水网络预测工具的涌现,其中提出的许多问题正在获得新的答案。但其核心命题——理解焓与熵的物理意义,而非仅仅优化一个数字——依然是现代药物设计中最值得深思的方法论议题之一。
参考文献(原文引用,节选核心)
- 1. Olsson, T. S. G., Williams, M. A., Pitt, W. R. & Ladbury, J. E. The thermodynamics of protein-ligand interactions and solvation: insights for ligand design. J. Mol. Biol.384, 1002–1017 (2008).
- 2. Biela, A. et al. Ligand binding stepwise disrupts water network in thrombin: enthalpic and entropic changes reveal classical hydrophobic effect. J. Med. Chem.55, 6094–6110 (2012).
- 3. Baum, B. et al. Non-additivity of functional group contributions in protein-ligand binding: a comprehensive study by crystallography and isothermal titration calorimetry. J. Mol. Biol.397, 1042–1057 (2010).
- 4. Biela, A. et al. Dissecting the hydrophobic effect on the molecular level: the role of water, enthalpy, and entropy in ligand binding to thermolysin. Angew. Chem. Int. Ed.52, 1822–1828 (2013).
- 5. Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today13, 869–874 (2008).
- 6. Ladbury, J. E., Klebe, G. & Freire, E. Adding calorimetric data to decision making in lead discovery: a hot tip. Nat. Rev. Drug Discov.9, 23–27 (2010).
- 7. Chodera, J. D. & Mobley, D. L. Entropy-enthalpy compensation: role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys.42, 121–142 (2013).
- 8. Steuber, H., Heine, A. & Klebe, G. Structural and thermodynamic study on aldose reductase. J. Mol. Biol.368, 618–638 (2007).
- 9. Brandt, T. et al. Congeneric but still distinct: how closely related trypsin ligands exhibit different thermodynamic and structural properties. J. Mol. Biol.405, 1170–1187 (2011).
- 10. Krimmer, S., Betz, M., Heine, A. & Klebe, G. Methyl, ethyl, propyl, butyl: futile but not for water. ChemMedChem9, 833–846 (2014).
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号