为什么90%的新药临床开发都失败了?

为什么90%的新药临床开发都失败了?

DrugIntel
发布于 2026-05-08 19:23:22
发布于 2026-05-08 19:23:22

文献来源: Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharmaceutica Sinica B. 2022;12(7):3049–3062. DOI: 10.1016/j.apsb.2022.02.002 作者单位: 密歇根大学药学院;百时美施贵宝公司(BMS)临床药理部
导读:一个令人不安的事实
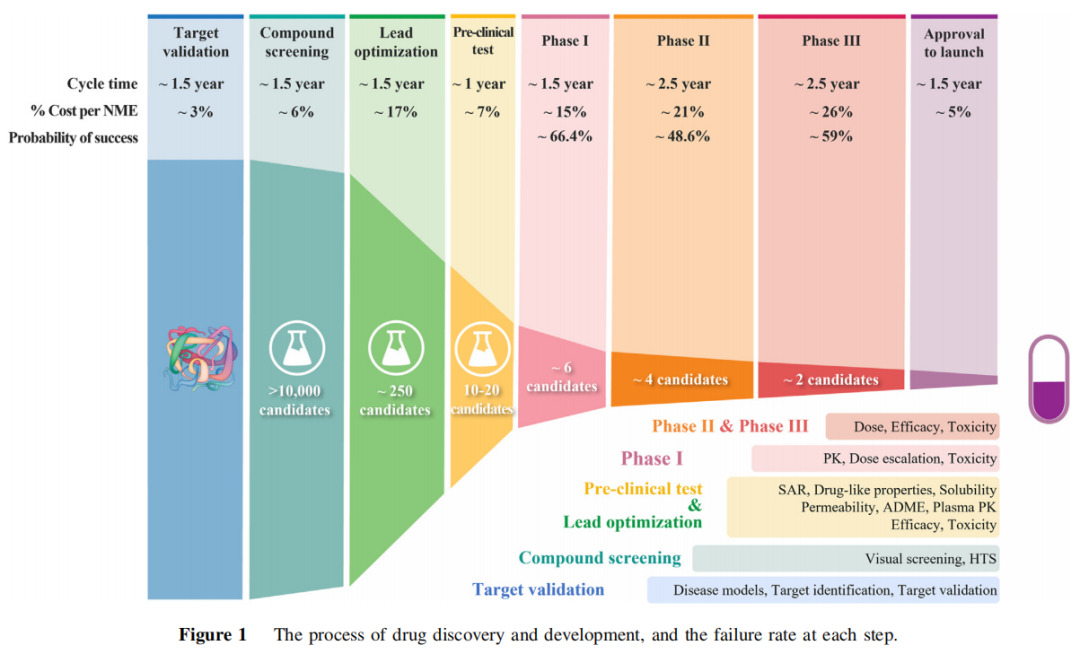
一款新药从靶点验证到上市,平均耗时 10–15年,花费逾 10–20亿美元。然而,即便是已经通过层层严格筛选、成功进入 I 期临床试验的候选药物,仍有约90%最终以失败告终。
更令人警醒的是:这90%的失败率,仅统计了进入临床阶段的候选药物。若将临床前阶段的淘汰纳入计算,真实失败率远不止于此。
过去几十年,学术界和工业界在药物研发的每一个环节——靶点验证、高通量筛选、AI辅助设计、药代动力学(PK)优化、临床试验设计——都投入了巨大资源并取得了显著进步,但总体临床成功率始终徘徊在 10%–15%,几乎毫无改善。
这篇来自密歇根大学团队的综述,直击这一核心困境,提出了一个长期被制药界系统性忽视的根源性问题,并给出了具体的改进框架。
一、临床失败的四大原因——现有认知的局限
基于2010–2017年临床试验数据的系统分析,临床失败的原因可归结为以下四类:
失败原因 | 占比 |
|---|---|
缺乏临床疗效(Lack of efficacy) | 40%–50% |
毒性难以管理(Unmanageable toxicity) | 30% |
药物特性不佳(Poor drug-like properties) | 10%–15% |
商业战略问题(Poor strategic planning) | ~10% |
值得注意的是:
- • 临床"缺乏疗效"并非意味着药物本身无效,而是候选药物在最大耐受剂量(MTD)下无法在病变靶器官中达到足够的药效浓度;
- • 临床"毒性不可控"的本质,往往是药物在正常健康器官中的暴露量过高,在达到治疗效果之前便已引发严重毒副作用;
- • 药物特性(ADME/PK)相关失败在过去20年已从30%–40%下降至10%–15%,证明优化策略有效——但整体成功率并未同步提升,说明真正的瓶颈在别处。
二、现行药物优化体系——做对了什么,又遗漏了什么?
2.1 做对了什么
当前药物优化流程已高度成熟,核心包括:
靶点验证层面:
- • 利用遗传学、基因组学、蛋白质组学在细胞系、动物模型及人类疾病模型中系统验证靶点;
- • 使用 siRNA 敲降、CRISPR 基因编辑确认靶点与疾病的因果关系;
- • AI/机器学习辅助靶点结构预测(如 AlphaFold)与虚拟筛选。
药物优化层面(SAR):
- • 通过结构–活性关系(Structure-Activity Relationship, SAR)将化合物对靶点的亲和力优化至低 nmol/L 甚至 pmol/L 级别(Ki 或 IC₅₀);
- • 筛选 hERG 抑制、激酶选择性谱、脱靶毒性等多维度安全性指标。
药物特性优化层面:
- • 遵循 Lipinski "五规则":分子量 <500 Da,氢键供体 <5,氢键受体 <10,cLogP <5;
- • 极性表面积(PSA)<140 Ų;体外渗透性 >2–3 × 10⁻⁶ cm/s;
- • 严格的体内 PK 筛选标准:生物利用度 F >30%,半衰期 t₁/₂ >4–6 h,清除率 CL <25% 肝血流量。
临床试验设计层面:
- • 基因组生物标志物指导的患者分层;
- • 精准的剂量爬坡设计;
- • I期–III期试验终点的精细化设计。
2.2 系统性遗漏了什么
尽管以上策略全面而严谨,作者指出:整个优化流程存在一个根本性盲点——对药物在"病变靶器官 vs. 正常器官"中暴露量与选择性的系统评估被长期忽视。
临床剂量/疗效/毒性的平衡,不仅取决于药物对靶点的效力与特异性(SAR),同样取决于药物在病变组织与健康组织间的暴露量与选择性分布(STR)——而后者在当前药物优化标准流程中几乎是缺席的。
三、核心问题:组织暴露/选择性的系统性忽视
3.1 "自由药物假说"的局限
当前以血浆 PK 为核心的候选药物筛选标准,其理论基础是"自由药物假说(Free Drug Hypothesis)":
- • 仅游离(未与蛋白结合)的药物能够分布进入组织;
- • 稳态时,血浆中游离药物浓度 ≈ 靶组织中游离药物浓度;
- • 因此,血浆中的药物暴露量可作为靶组织药效的替代指标。
然而,该假说在以下情形下并不成立(甚至完全相反):
- 1. 主动转运体的存在:肝脏、脑等器官中存在大量摄取与外排转运体(如 OATP、P-gp、BCRP),可造成药物在血浆与组织之间呈不对称分布;
- 2. 离子化药物的"溶酶体截留"效应:弱碱性药物在酸性溶酶体中蓄积;
- 3. 白蛋白的主动转运:白蛋白本身可经 FcRn 等主动转运途径从循环转运至组织,携带结合型药物至特定组织;肿瘤微环境中的 SPARC 蛋白可促进白蛋白-药物复合物在肿瘤组织中的蓄积;
- 4. 共价结合、前药代谢、蛋白降解剂(PROTAC)等新型机制;
- 5. 局部给药(如吸入给药)的特殊 PK 行为。
结论: 血浆暴露量高的候选药物,其在病变靶器官中的实际暴露量可能严重不足;而血浆暴露量看似偏低的候选药物,可能恰恰是因为大量药物分布进入了靶组织——这两类药物在现行筛选体系中会得出截然相反的错误结论。
3.2 临床案例佐证
EGFR 抑制剂家族(吉非替尼、拉帕替尼、厄洛替尼、凡德他尼): 尽管共享相似的药效团和接近的 SAR/PK 特性,四者在不同癌种中的临床疗效与毒性谱却差异显著。仅凭 SAR 和药物特性无法解释这些差异,结构微调对其在不同肿瘤组织中的分布选择性可能是关键因素。
BTK 抑制剂(伊布替尼、阿卡替尼、泽布替尼 vs. spebrutinib): 前三者均获批上市,而 spebrutinib 在早期临床中折戟。除靶点特异性差异外,各药物在病变淋巴组织 vs. 心脏等正常器官中的暴露量差异,可能是决定其临床命运的重要维度。
选择性雌激素受体调节剂(SERMs): 超过600项 SERM 临床试验,11个已获批(他莫昔芬、雷洛昔芬等),众多结构高度相似的候选药物却在乳腺癌、骨质疏松、更年期症状等不同适应症中呈现截然不同的疗效/毒性谱。研究表明,微小的结构修饰会显著改变药物在肿瘤、脂肪垫、骨骼等组织中的暴露量与选择性,进而决定临床结局。
瑞德西韦(Remdesivir)治疗 COVID-19: 尽管体外抗 SARS-CoV-2 活性良好,100 mg 静脉给药方案的临床疗效十分有限——作者分析认为,其在肺组织(靶器官)的暴露量/选择性可能不足以充分抑制病毒,同时在肾脏(毒性靶器官)的暴露量却相对偏高,形成了不利的剂量/疗效/毒性平衡。
四、STAR 框架:整合组织暴露维度的新型药物优化体系
4.1 框架定义
作者提出结构–组织暴露/选择性–活性关系(Structure-Tissue exposure/selectivity-Activity Relationship, STAR)框架,在现有 SAR + 药物特性评估体系的基础上,引入组织暴露/选择性(STR,Structure-Tissue exposure/selectivity Relationship)作为第三维度,将候选药物分为四类:
组织暴露/选择性(STR)
低 高
┌──────────┬──────────┐
高 │ Class II │ Class I │
效力/特异性 │ 高剂量需求 │ 低剂量需求 │
(SAR) │ 毒性风险高 │ 疗效优越 │
├──────────┼──────────┤
低 │ Class IV │ Class III│
│ 应终止开发 │ 潜力被低估│
│ │ 毒性可控 │
└──────────┴──────────┘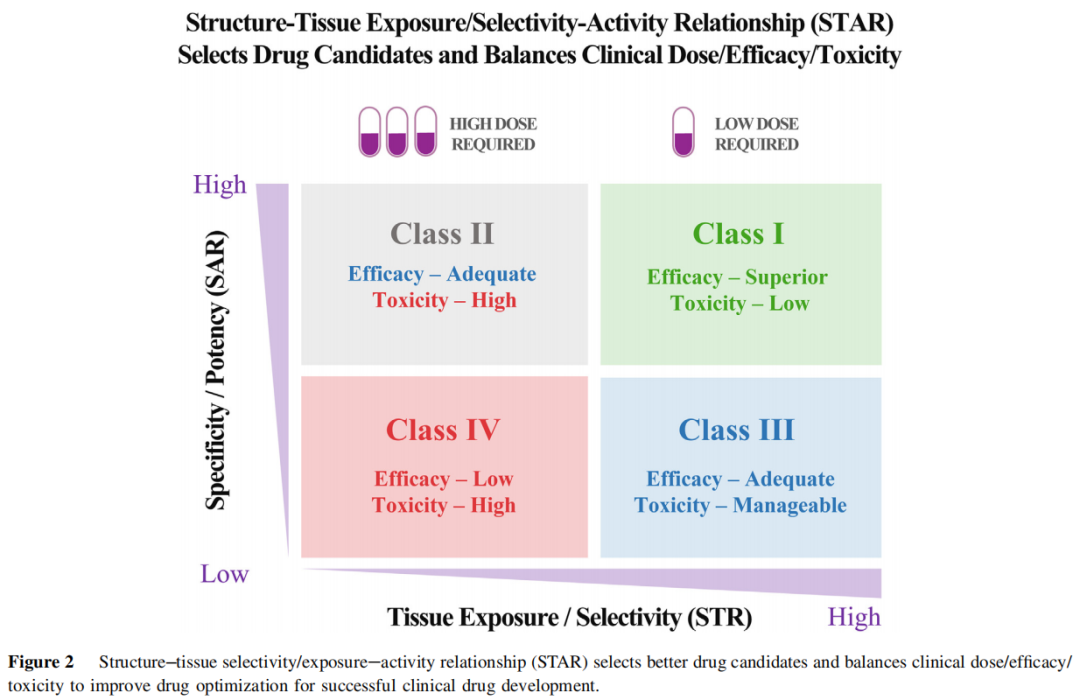
4.2 四类药物详解
Class I(高 SAR + 高 STR)——最理想,成功率最高
- • 对靶点效力强(Ki < 数百 nmol/L),同时在病变组织中高度富集;
- • 低剂量即可实现优越疗效,健康器官暴露量低,安全性好;
- • 代表药物(推测):索非布韦(NS5B抑制剂)、阿托伐他汀(HMG-CoA还原酶抑制剂)、西地那非(PDE5抑制剂)、阿卡替尼(BTK抑制剂)。
- • 注:部分 Class I 药物可能在靶器官和某些正常器官中均有高暴露(如多柔比星、他莫昔芬),表现为高疗效伴随较高不良事件发生率。
Class II(高 SAR + 低 STR)——需谨慎评估
- • 体外效力优秀,但靶组织分布选择性差;
- • 为达到疗效往往需要高剂量,但高剂量又导致在正常器官的毒性积累,形成"剂量陷阱";
- • 这是当前药物优化最集中投入的类别,也是最容易陷入"体外很完美,临床频失败"困境的类别;
- • 一个典型现象:化合物体外 IC₅₀ 在 nmol/L 级别,但体内有效浓度需达 μmol/L——中间数量级的鸿沟,正是组织暴露不足的体现;
- • 代表药物(推测):伊布替尼、联合非达他汀(Fedratinib)、瑞德西韦(若以低nmol/L IC₅₀计)。
Class III(低 SAR + 高 STR)——最常被错误淘汰的类别
- • 体外效力相对较低(几百 nmol/L < Ki < 1 μmol/L),但在病变组织中高度选择性富集;
- • 低至中等剂量即可实现充分疗效,正常器官暴露量低,毒性可控;
- • 在现行以血浆 PK 为核心的筛选体系中,这类药物因血浆暴露量低而被系统性淘汰——而血浆暴露量低,恰恰可能是因为药物大量分布进入了靶组织;
- • 注意:IC₅₀ 仍需 <1 μmol/L;若效力过低(IC₅₀ >数 μmol/L),即使组织选择性优异也难以达效;
- • 代表药物(推测):沙利度胺(E3连接酶抑制剂,作为泛免疫调节剂)。
Class IV(低 SAR + 低 STR)——应尽早终止
- • 效力低,靶组织选择性差;
- • 高剂量下毒性不可控,疗效依然不足;
- • 应在早期优化阶段即予以淘汰,避免资源浪费。
五、STR 的测量方法与实施路径
5.1 核心测量参数
STR 可通过以下参数量化:
组织暴露量(AUC):
组织血浆
其中 (血浆分配系数)= 组织总药浓 / 血浆总药浓(或 AUC 之比)。
组织选择性(两种计算方式):
- • 方式一(适用于毒性靶器官未知时):
组织选择性组织所有组织
全面评估所有主要器官的药物分布,用于识别潜在毒性靶器官,适合小批量候选药物深度分析。
- • 方式二(适用于毒性靶器官已知时):
组织选择性(比值)效应组织()毒性组织()
直接评估治疗窗,效率更高,适合候选药物的比较性筛选。
关于"总药物浓度 vs. 游离药物浓度"的争议:
作者倾向于使用总药物浓度/暴露量进行 STR 评估,理由是:游离药物浓度极难准确测量(组织匀浆法破坏亚细胞结构,无法真实反映作用位点的游离浓度),且 AstraZeneca 的 PK/PD 模型研究表明,总药物水平与临床 PK/PD 关系的相关性优于游离药物水平。
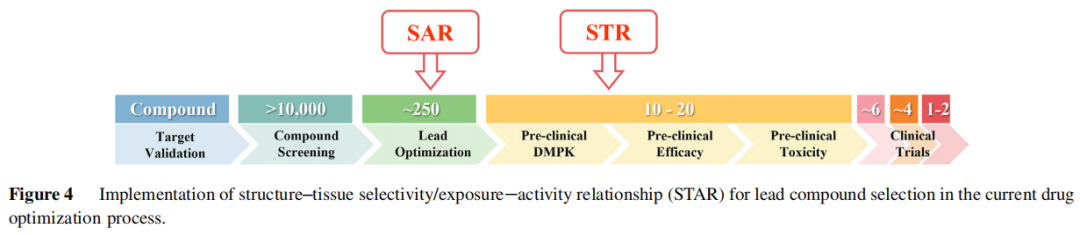
5.2 在药物优化流程中的嵌入策略
作者建议将 STR 评估作为现有筛选流程的补充环节而非颠覆性替代,具体路径如下:
>10,000 化合物
↓ 体外活性筛选(SAR,IC₅₀/Ki)
~250 化合物
↓ 体外 ADME 筛选(溶解度/渗透性/稳定性/蛋白结合)
10–20 化合物
↓ 体内血浆 PK 评估
少数候选药
↓ 【新增】体内组织暴露/选择性评估(STR)
↓ 综合 SAR + ADME + 血浆 PK + 组织 STR 做最终决策
临床候选药(1–2 个)关键点在于:STR 的体内评估资源消耗大,不适合对数百个化合物全面铺开,应在已完成血浆 PK 筛选后,仅对少数进入候选阶段的化合物实施。
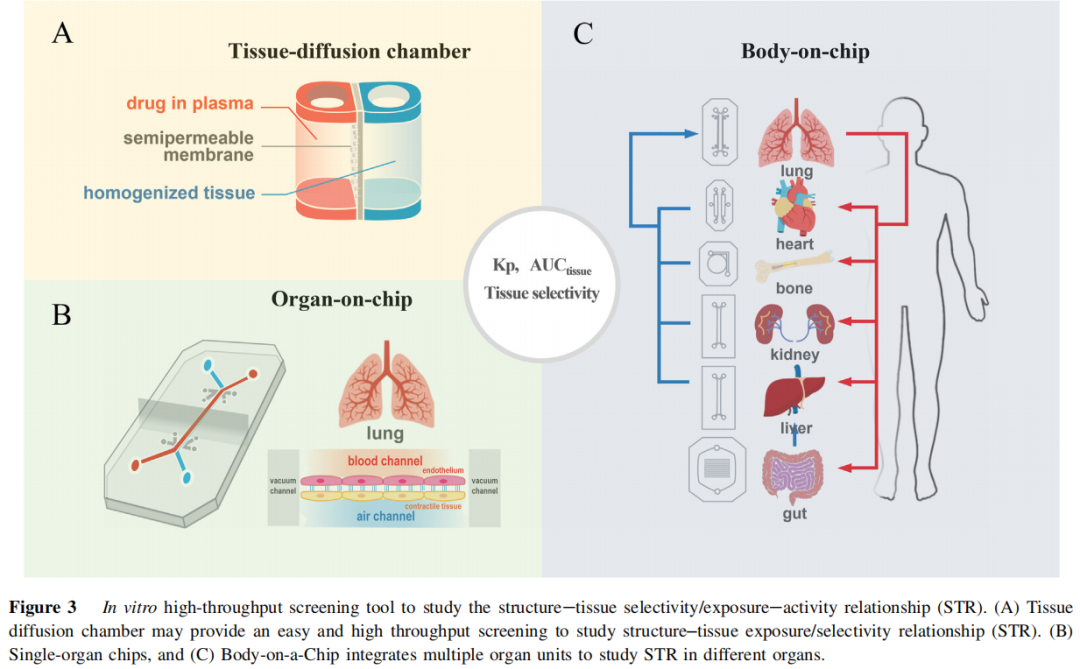
5.3 技术实现路径(未来方向)
体外高通量 STR 筛选:
- • 组织扩散室(Tissue Diffusion Chamber):模拟体循环与各组织间的药物分配,通过半透膜将"血浆室"与"匀浆组织室"分隔,测定 及组织选择性,可实现较高通量;
- • 器官芯片(Organ-on-a-Chip):整合人体生理条件下的血管/上皮屏障及实质细胞,更精准模拟药物跨组织屏障的分布行为;
- • 多器官芯片(Body-on-a-Chip):整合多个器官单元,模拟全身药物分布,为 STR 提供更全面的体外预测平台。
AI 辅助计算预测:
- • 利用已有 STR 数据,构建化学描述符(logP、拓扑参数、蛋白结合、Kappa 形状指数等)与组织 、选择性之间的机器学习预测模型;
- • FDA 新药申请(NDA)数据库中包含大量已批准药物的 ¹⁴C 质量平衡数据、组织分布数据及临床疗效/毒性数据,若能整合用于模型训练,将极大加速 AI-STR 预测能力的建立;
- • 此举需要学术界、FDA 与工业界的协同合作。
生理药代动力学模型(PBPK): 现有 PBPK 模型主要用于药物相互作用预测和儿科/首次人体给药剂量预测,其预测临床组织暴露量的能力受限于:(1)依赖大量动物组织浓度数据,耗时耗力;(2)临床阶段人体组织浓度数据难以获取,模型无法实时校正。未来若能结合体外 STR 筛选数据驱动 PBPK 建模,将显著提升预测精度和实用性。
临床阶段非侵入性影像技术:
- • 动态 PET 成像:利用放射性标记化合物可视化体内药物分布及靶点占有率;已有案例显示 PET 成功指导 NK1 受体拮抗剂的剂量优化及开发决策;但常用正电子发射核素(¹¹C:20 min;¹⁸F:110 min)半衰期短,且 ¹⁸F 标记可能改变药物本身的组织分布特性;
- • 质谱成像(MSI):目前仅能对离体组织切片进行药物分子可视化,手持式体内探针尚在研发中;
- • 总体而言,适用于 STAR 临床研究的成像技术目前仍有较大缺口,亟待新技术突破。
六、对靶点验证的再认识
值得强调的是,STAR 框架的核心并非否定靶点验证的重要性,而是在承认其局限性的基础上提出补充方案。
靶点验证本身面临两大根本挑战:
- 1. 靶点与疾病因果关系的验证:体外细胞系、动物模型与人类疾病之间的生物学差异,使得真正意义上的靶点验证只能在首个成功药物出现后才能实现——这是"先有鸡还是先有蛋"的困境,尤其限制了 first-in-class 药物的开发;
- 2. 药物与靶点结合的验证:部分已成功上市的药物,其实际药理机制与最初设计时的靶点并不一致——这意味着基于"错误靶点"的 SAR 优化可能从源头就走偏了方向。
即便靶点验证完全正确,仍有大量候选药物在临床失败——这正是 STAR 框架所要解决的核心问题所在。
七、STAR 体系的现实意义与局限
预期收益
作者估计,若 STAR 体系得到广泛应用,临床药物开发成功率有望从当前的 10%–15% 提升至 30%–40%。这一改善,意味着:
- • 数十亿美元的研发资源从注定失败的项目中解放出来;
- • 更多有效药物能够更快地到达患者;
- • Class III 潜力候选药的系统性发掘。
重要局限与未解问题
- • 分类标准尚未量化:目前文中所有药物分类示例均为推测性评估,缺乏实验性 STR 数据的支撑,"高/低组织暴露"的切分阈值尚未被定义;
- • 方法学体系仍不完善:STR 测量方法(组织扩散室、器官芯片等)尚处于概念和早期研究阶段,未形成标准化、高通量的工业应用规程;
- • STAR 并非万能:靶点验证不足、战略规划失误等问题不在 STR 优化的覆盖范围内,STAR 是必要补充,但不是"银弹";
- • 总药物浓度 vs. 游离药物浓度的争论仍未有定论,后者虽更贴近机制,但准确测定极为困难。
写在最后
这篇文章的价值,不在于提出了一个全新的实验技术,而在于系统性地揭示了制药行业长期内化的一个认知偏差——我们用血浆 PK 参数代替组织暴露评估,不是因为这样做是对的,而是因为这样做最方便,且长期以来没有人追问这个假设。
从方法论层面看,STAR 框架本质上是将治疗指数(Therapeutic Index)的概念从"血浆浓度空间"拓展至"组织浓度空间",是对传统 PK/PD 框架在药物优化应用中的一次重要修正。
从实践层面看,Class III 候选药物的"平反"尤为值得关注。在日益激烈的靶点竞争中,那些被主流筛选体系淘汰的"低血浆暴露"化合物,很可能藏匿着真正具有临床价值的候选药——这是一个被系统性忽视的机会。
当然,将 STR 研究真正纳入工业化药物优化流程,仍面临巨大的技术、成本和数据挑战。但如本文所言:若能将成功率从10%提升至30%,这将是整个药物研发领域有史以来最具影响力的系统性改进之一。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号