Nature | 基于稀疏数据的可迁移对映选择性预测模型

Nature | 基于稀疏数据的可迁移对映选择性预测模型

DrugAI
发布于 2026-03-03 17:27:12
发布于 2026-03-03 17:27:12
DRUGONE
在不对称催化研究中,如何预测反应的对映选择性一直是核心挑战之一。传统方法通常依赖大量实验数据进行建模,但在真实化学研究中,可获得的数据往往高度稀缺,导致模型难以推广到新的底物、催化剂或反应体系。研究人员提出了一种能够从稀疏数据中学习并实现跨体系迁移的对映选择性建模策略,通过将物理有意义的分子表示与机器学习方法相结合,使模型能够在极少实验样本条件下准确预测新的不对称反应结果。该方法展示了在多个反应家族之间的可迁移性,为利用小数据驱动催化发现提供了一条可行路径。

对映选择性的精准控制是现代合成化学的重要目标,尤其在药物分子构建中,不同对映体往往表现出完全不同的生物活性。然而,对映选择性来源于复杂的立体电子相互作用,其影响因素包括催化剂结构、底物构型、非共价相互作用以及反应环境等多重因素,使得经验规则难以普适适用。虽然机器学习近年来被用于预测选择性,但大多数模型需要密集实验数据进行训练,并且通常局限于单一反应体系,一旦条件变化便难以泛化。因此,建立能够在“小数据”条件下仍具预测能力、并可迁移至新体系的模型,是推动数据驱动不对称催化的重要问题。
稀疏数据驱动的建模策略
研究人员提出将化学知识嵌入模型构建过程,而非完全依赖数据规模。通过设计能够反映立体效应与电子效应的分子描述符,模型可以在有限样本中捕捉决定对映选择性的关键物理因素。与传统黑箱学习不同,这种方法强调使用可解释的特征空间,使模型在训练数据极少的情况下仍能建立稳定的结构–选择性关联,从而避免对大规模实验数据的依赖。
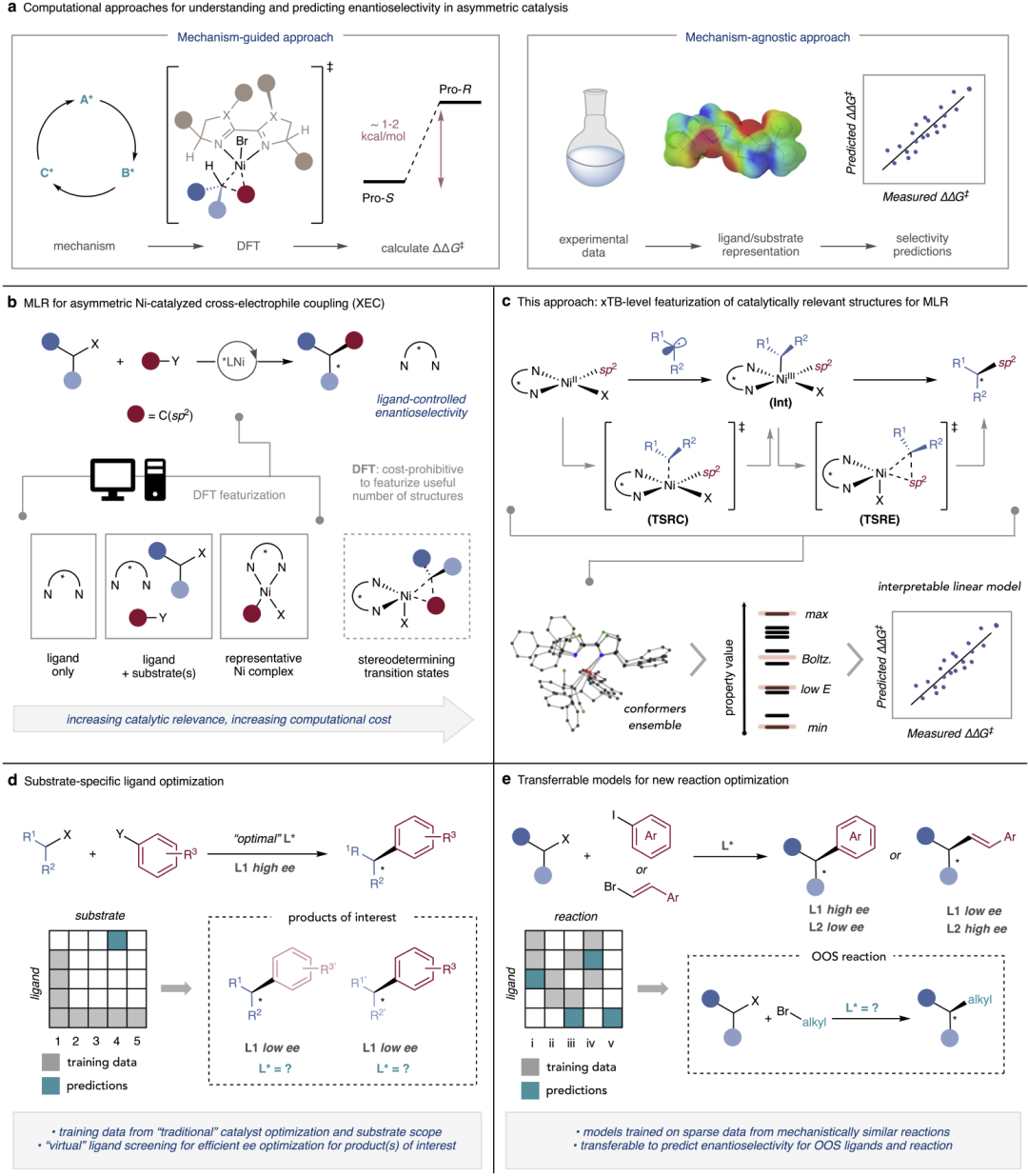
图1:稀疏数据条件下的对映选择性建模概念框架。
跨反应体系的可迁移学习
在模型训练完成后,研究人员将其应用于不同但相关的反应体系,以测试其迁移能力。结果表明,模型不仅能够重现训练体系中的选择性趋势,还能在未见过的新底物和催化剂组合中保持良好预测性能。这说明模型学习到的是更一般性的立体控制规律,而非简单的数据拟合。通过这种方式,小规模实验数据即可支持对更广泛化学空间的探索。
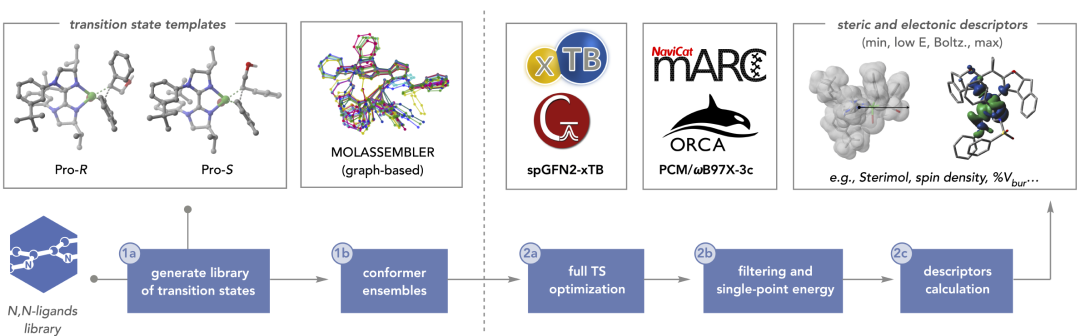
图2:模型在不同反应家族之间的迁移预测表现。
物理可解释性与选择性来源解析
进一步分析显示,模型识别出的关键特征与已知的化学直觉高度一致,例如空间位阻分布和非共价作用对过渡态稳定性的影响。这种一致性说明模型不仅具备预测能力,还能够作为分析工具帮助理解对映选择性来源,为催化剂优化提供理论指导。
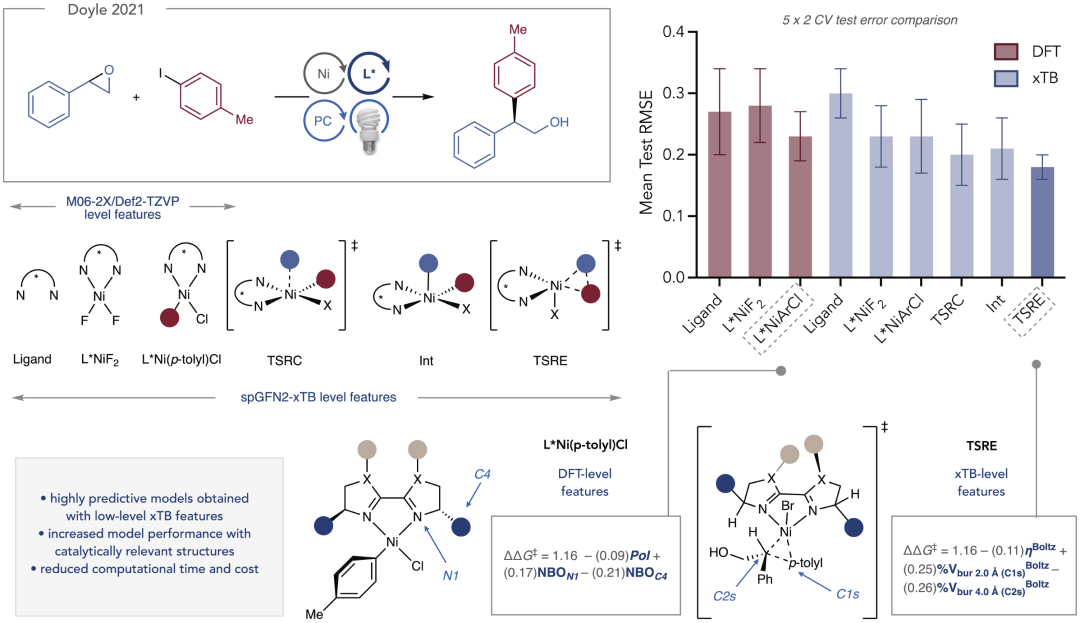
图3:模型特征与立体电子效应的关联分析。
在小样本条件下的预测能力验证
研究人员通过逐步减少训练数据量来测试模型的稳健性。即便在仅使用少量实验结果的情况下,模型仍然能够保持较高预测准确度,显著优于依赖大数据训练的传统机器学习方法。这一结果验证了该策略在真实实验资源受限场景中的实用价值。
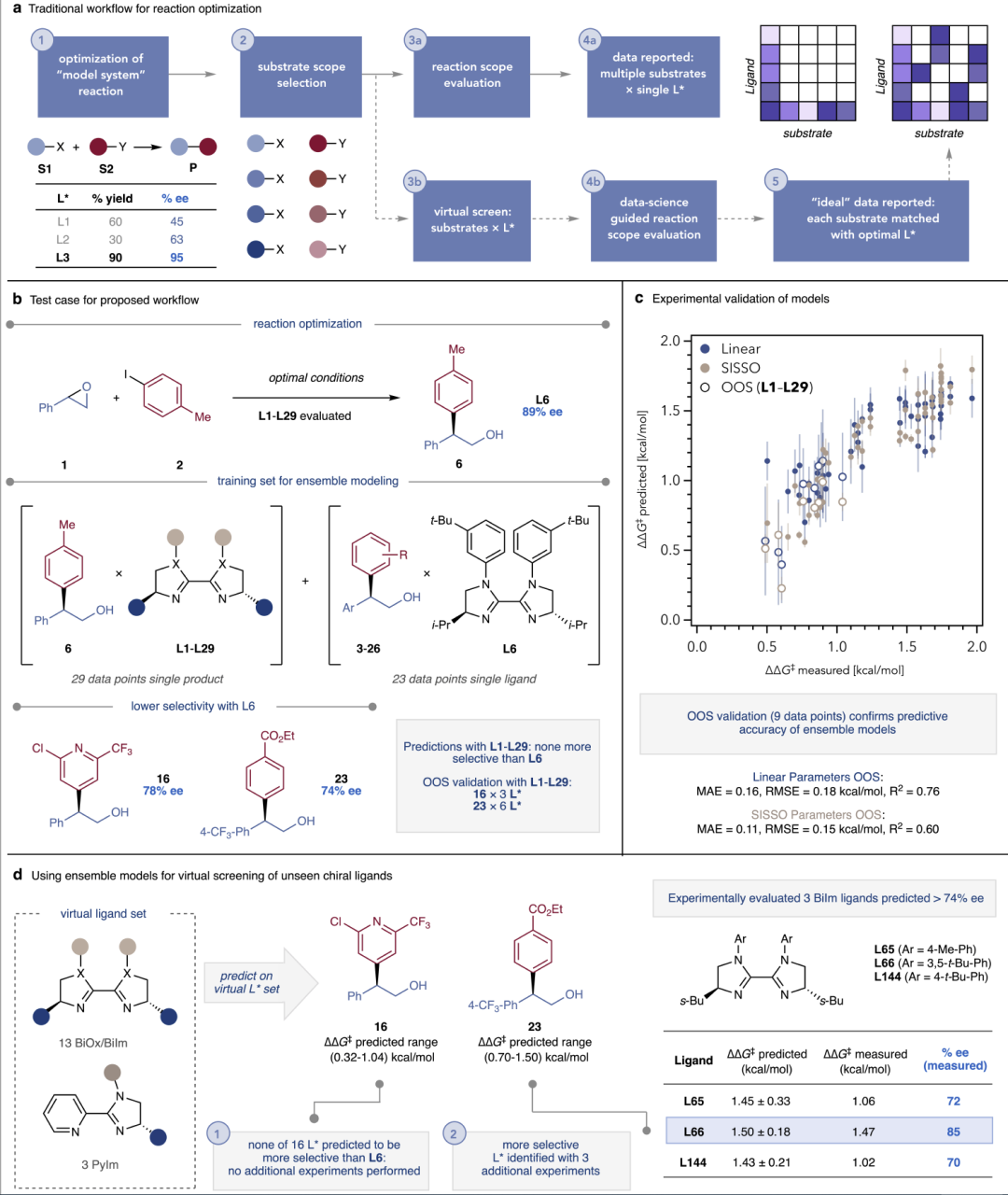
图4:不同数据规模下模型预测性能对比。
面向催化发现的应用潜力
该模型可用于指导新催化剂和新底物组合的快速筛选,从而减少盲目实验尝试。通过预测潜在高选择性反应条件,研究人员能够优先开展最有希望的实验,大幅提高研究效率。这种数据与机理相结合的建模方式,为不对称催化从经验驱动走向数据驱动提供了新的工具。
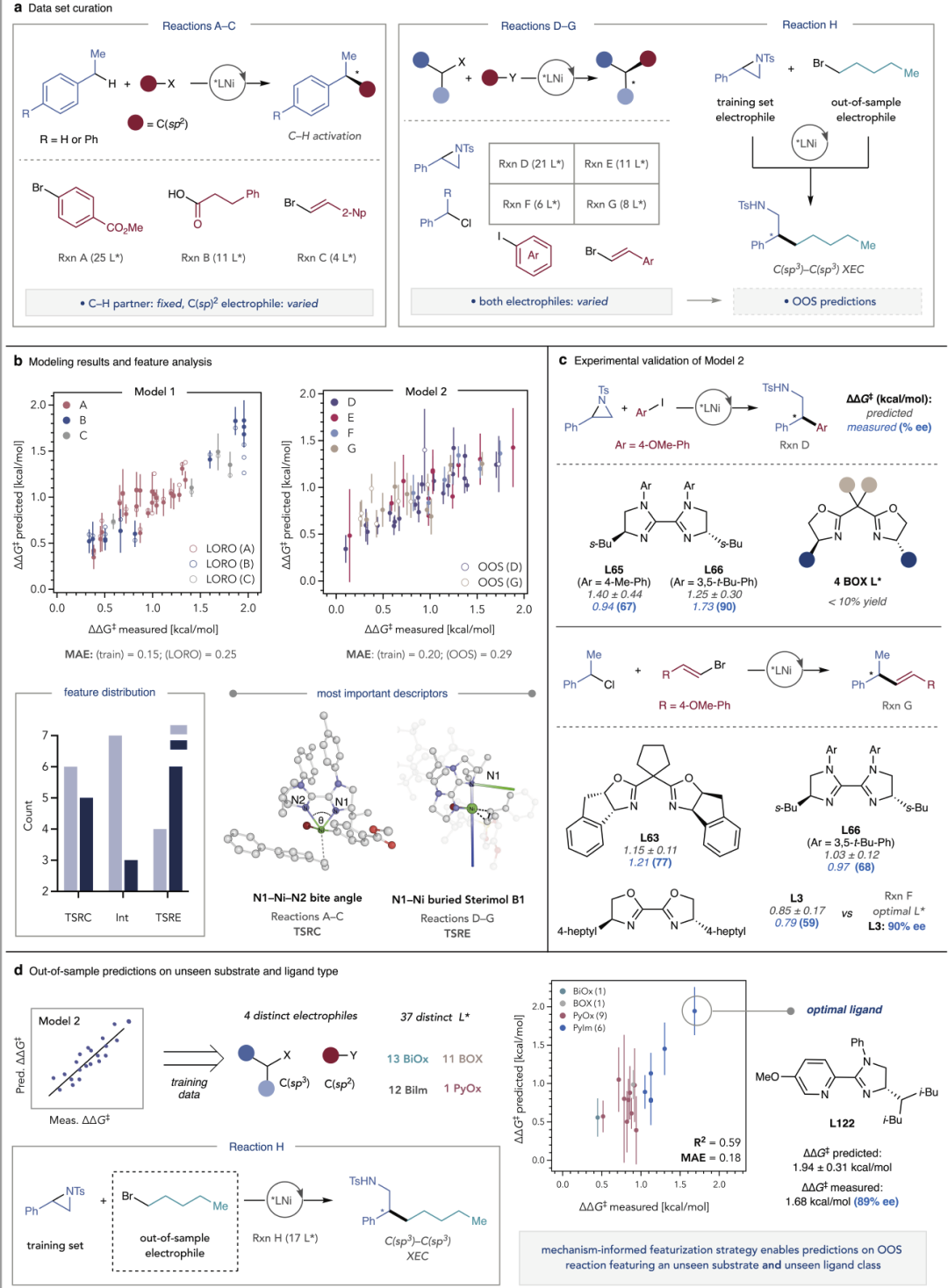
图5:模型辅助催化剂与反应条件优化示意。
方法意义与未来展望
研究人员展示了一种不同于“大数据训练”的科学机器学习范式,即通过嵌入化学机理知识,使模型能够在数据稀缺条件下实现可靠预测并具备跨体系迁移能力。未来,这一策略有望扩展到更多类型的选择性问题,如区域选择性、构型选择性以及复杂反应网络建模,并与自动化实验平台结合,形成闭环的智能催化发现体系。
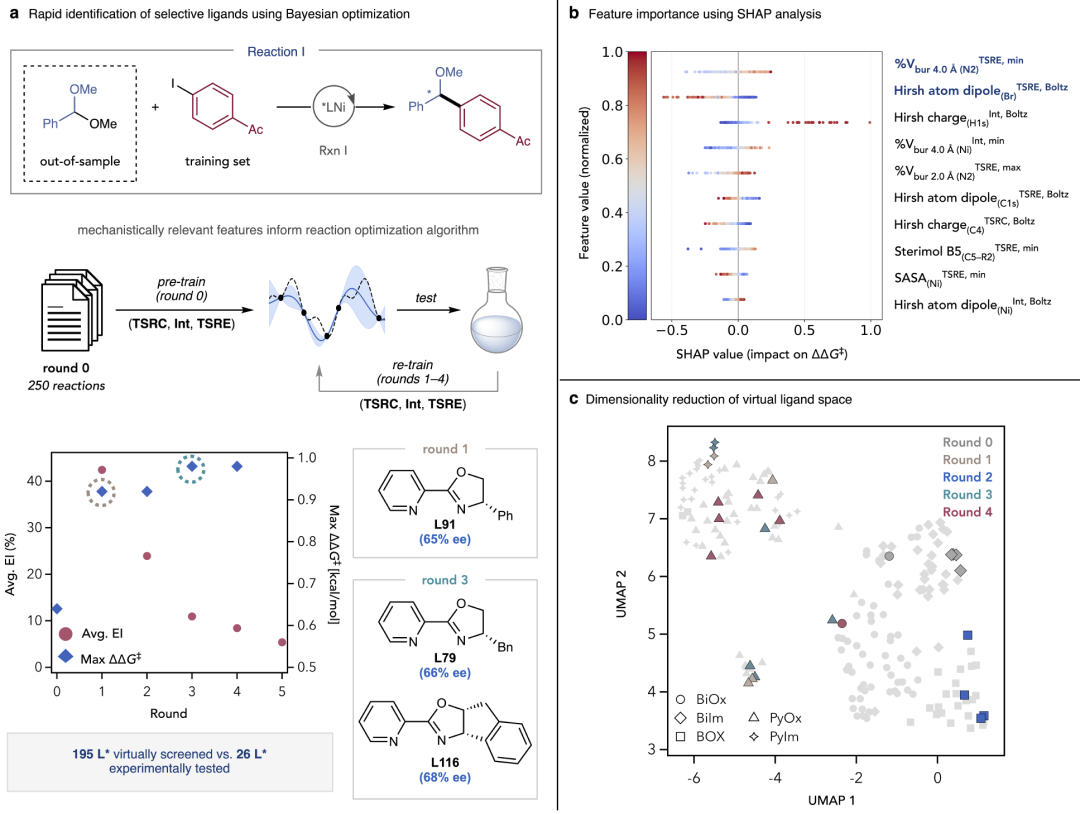
图6:稀疏数据驱动的可迁移选择性建模在未来化学研究中的应用前景。
参考资料
Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature (2026).
https://doi.org/10.1038/s41586-026-10239-7

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-02-12,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号