J. Med. Chem. | 通过计算机辅助药物设计发现高效非共价型 SARS-CoV-2 主蛋白酶抑制剂 !

J. Med. Chem. | 通过计算机辅助药物设计发现高效非共价型 SARS-CoV-2 主蛋白酶抑制剂 !

DrugOne
发布于 2025-11-17 20:36:43
发布于 2025-11-17 20:36:43

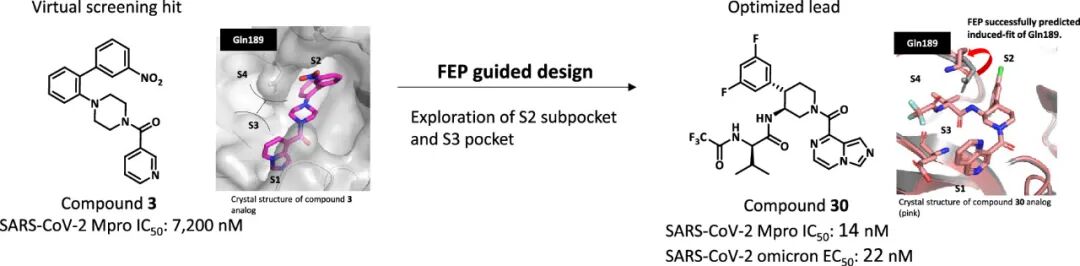
自COVID-19疫情全球大流行以来,SARS-CoV-2的持续变异(如奥密克戎及其亚分支)对现有治疗体系构成持续挑战。尽管以奈玛特韦、恩赛特韦为代表的主蛋白酶(Mpro)抑制剂已成为临床核心治疗药物,但其共价结合特性带来的潜在毒性、对CYP450酶系的抑制引发的药物相互作用,以及对其他冠状病毒覆盖不足的局限性日益凸显。近日,发表于《Journal of Medicinal Chemistry》的研究通过计算机辅助药物设计(CADD)技术,成功开发出系列高活性非共价Mpro抑制剂,其中化合物30展现出超越现有药物的综合性能,为广谱抗冠状病毒药物研发树立了新标杆。本文将从靶点生物学价值、研发技术路线、候选药物性能及科学意义四个维度,深度解析这一成果。
一、靶点再认知:Mpro为何仍是抗冠状病毒药物研发的"黄金靶点"
SARS-CoV-2主蛋白酶(Mpro,又称3CLpro)作为病毒复制周期中的关键酶,其生物学特性决定了其不可替代的药物研发价值,这也是该研究坚持以其为靶点的核心逻辑。
1. 病毒复制的 不可替代枢纽
Mpro负责将病毒RNA翻译产生的多聚蛋白前体(pp1a/pp1ab)精准切割为11种非结构蛋白(nsp1-nsp11),这些蛋白共同构成病毒复制转录复合物(RTC)。与其他病毒酶(如RNA依赖的RNA聚合酶RdRp)相比,Mpro的切割位点具有高度特异性(识别Leu-Gln↓(Ser/Ala/Gly)序列),且切割过程无冗余机制——一旦Mpro活性被抑制,病毒复制将完全停滞,这为药物干预提供了明确的作用节点。
2. 跨冠状病毒的 结构保守性
序列比对显示,SARS-CoV-2 Mpro与SARS-CoV-1 Mpro的氨基酸序列同源性高达96%,与MERS-CoV Mpro的同源性也达76%,其催化活性中心(由His41、Cys145和Asn142组成)在所有已知人类冠状病毒中完全保守。这种结构保守性意味着针对该靶点的药物有潜力实现"广谱覆盖",避免因病毒变异导致的耐药性,这也是应对未来冠状病毒新发突发疫情的关键需求。
3. 人体的 低同源性 优势
人类基因组中不存在与Mpro同源的蛋白酶,这意味着Mpro抑制剂不易与人体蛋白发生交叉作用,从根本上降低了脱靶毒性风险。相比之下,RdRp抑制剂(如莫诺拉韦)可能影响人体线粒体RNA聚合酶,存在潜在的长期安全性隐患。
二、现有抑制剂及其局限性
目前,已有多种靶向Mpro的药物进入临床阶段,其中最具代表性的为奈玛特韦(nirmatrelvir, Paxlovid 的活性成分)和恩西特韦(ensitrelvir, S-217622)。 奈玛特韦是一种共价抑制剂,通过与Cys145形成可逆共价键来阻断Mpro活性。虽然其抗病毒效果显著,但共价抑制剂通常面临如下问题:
- 可能存在选择性不足与免疫原性风险;
- 对活性位点突变的耐受性较差,易受病毒变异影响;
- 在部分情况下可能导致不良药代动力学性质。
相比之下,非共价抑制剂通过可逆的非共价相互作用与靶标结合,具有更好的安全性、选择性及优化空间。代表性分子ensitrelvir已获日本批准上市,显示出良好的临床潜力。然而,其针对不同冠状病毒的活性仍存在一定局限性,这推动了研究者探索新的非共价化学骨架与设计策略。
三、理性设计的技术闭环:从虚拟筛选到先导化合物的全流程解析
该研究最显著的技术亮点是构建了"虚拟筛选-结构解析-FEP优化-成药性评价"的CADD闭环体系,将传统药物研发的 试错模式 转变为 精准设计模式 ,大幅提升了研发效率与成功率。
1. 双维度虚拟筛选:高效锁定初始Hit化合物
研究团队未采用传统高通量筛选(HTS),而是基于武田制药(Takeda)150万化合物库,实施了互补的双重虚拟筛选策略:
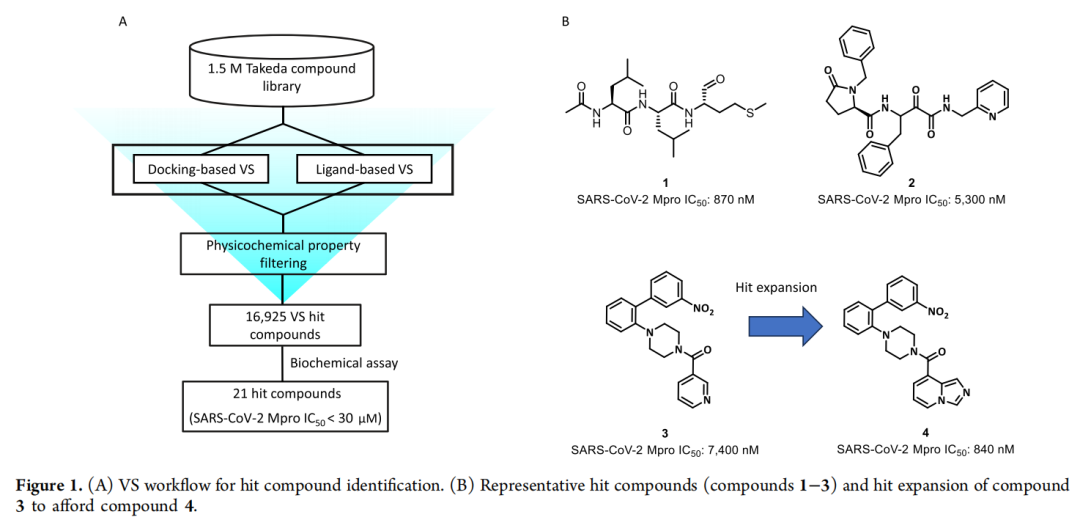
- 基于结构的虚拟筛选(SBVS):利用4个高分辨率SARS-CoV-2 Mpro晶体结构(PDB: 6LU7、6M03、6Y2G、5R7Y),通过分子对接(Glide SP)模拟化合物与Mpro活性口袋的结合模式。在非共价对接中,筛选 GlideScore ≤ −6.5 的化合物;在共价对接中,筛选 得分 ≤ −4.5 的化合物。物化性质筛选标准如下(使用 Pipeline Pilot 2017 版本执行):
- 1.分子量(MW):200 ≤ MW ≤ 600
- 2.分配系数(AlogP):−2 ≤ AlogP ≤ 5
- 基于配体的虚拟筛选(LBVS):以已报道的SARS-CoV-1 Mpro抑制剂为模板,采用3种相似性搜索方法:
- ECFP-4(扩展连接指纹)
- 基于拓扑药效团模型的 CATS 描述符(Chemically Advanced Template Search)
- 基于原子/键匹配的 MCS(最大公共子结构)搜索
通过=ADMET性质预筛,共获得16,925个虚拟筛选命中化合物(VS hits),随后在SARS-CoV-2 Mpro生化抑制实验中进行了活性测试。最终获得21个具有不同骨架类型的命中化合物,IC₅₀均低于30 μM。其中化合物3(哌嗪母核结构)表现出非共价抑制活性(SARS-CoV-2 Mpro IC₅₀=7200 nM),虽活性中等,但因结构新颖、无亲电活性基团,被确定为先导优化的 种子化合物。
2. 共晶结构引导的骨架重塑:突破广谱性瓶颈
为明确优化方向,研究团队解析了化合物4(化合物3的甲基化衍生物)与Mpro的复合物晶体结构(分辨率1.4 Å),通过电子密度图分析发现关键问题:
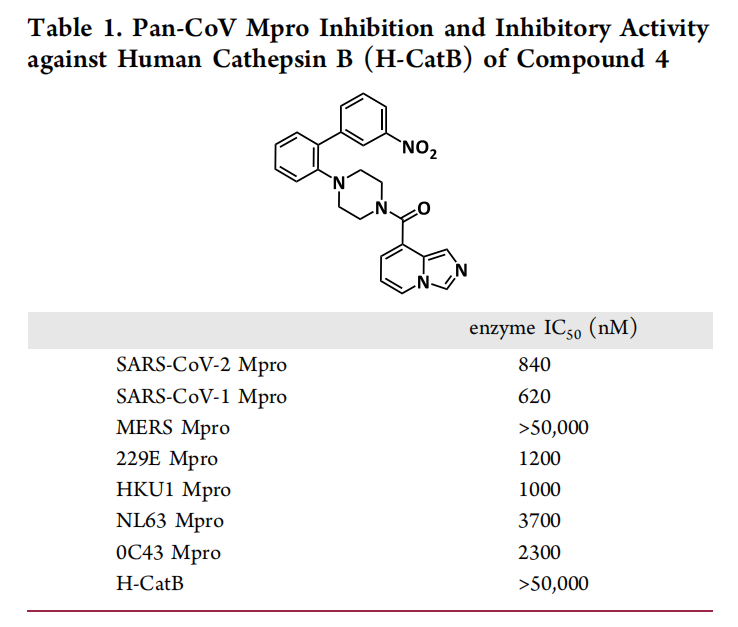
- 化合物4的硝基苯基基团与MERS-CoV Mpro的Leu49、Met25残基存在明显空间位阻,导致其对MERS-CoV Mpro抑制活性微弱(IC₅₀>50000 nM);
- Mpro活性口袋的S3亚口袋(由Gln189、Arg188组成)存在未被占据的疏水区域,可作为增强结合的潜在位点。

基于此,团队实施了两项关键结构改造:一是移除硝基苯基基团,消除空间冲突;二是将哌嗪母核重构为三取代哌啶母核,在P3位引入环己基,与S3口袋形成强疏水作用。
3. FEP计算驱动的 potency飙升:实现活性百倍提升
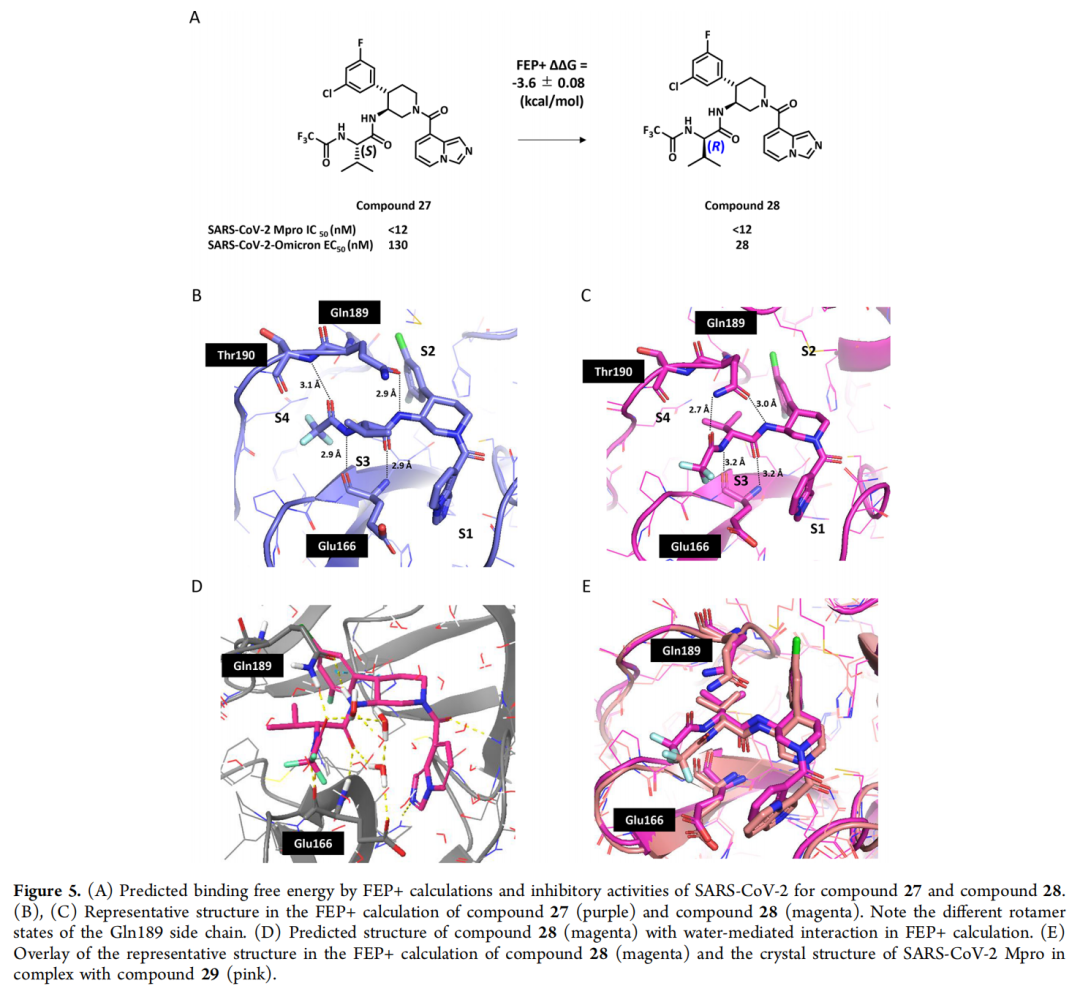
自由能微扰(FEP)计算作为该研究的 核心加速器,通过分子动力学模拟精准预测化合物与靶点的结合自由能,缓解了传统改造中 活性预测不准 的难题。研究团队针对S2和S3亚口袋实施了系统性FEP筛选:
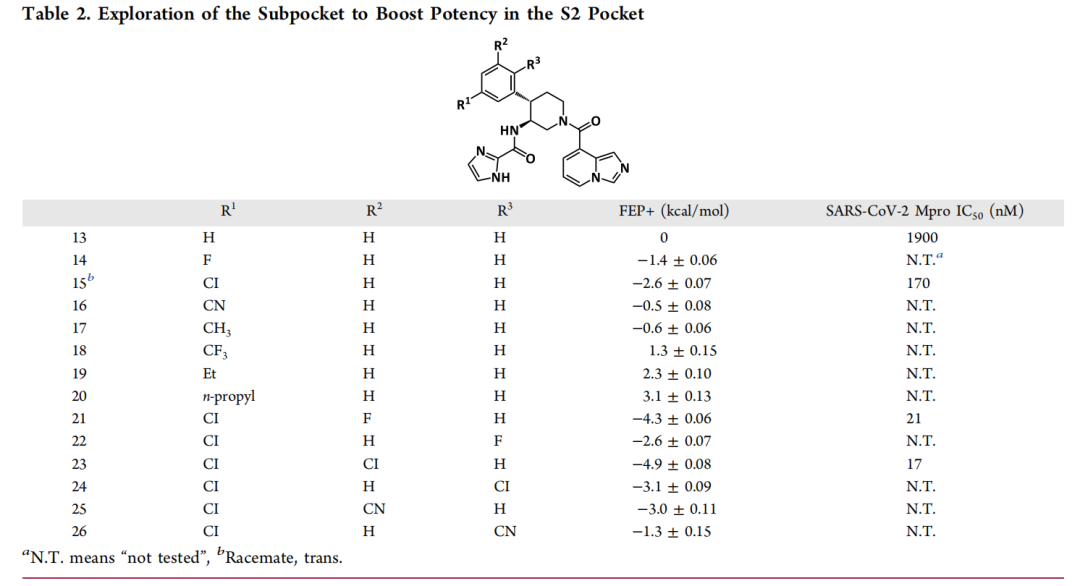
- S3口袋优化:预测在哌啶母核P3位引入3-氟苄基,可诱导Gln189残基发生构象变化,形成额外氢键。实验验证显示,该修饰使化合物对SARS-CoV-2 Mpro的IC₅₀从28000 nM降至950 nM,同时获得对MERS-CoV的抑制活性(IC₅₀=4200 nM);
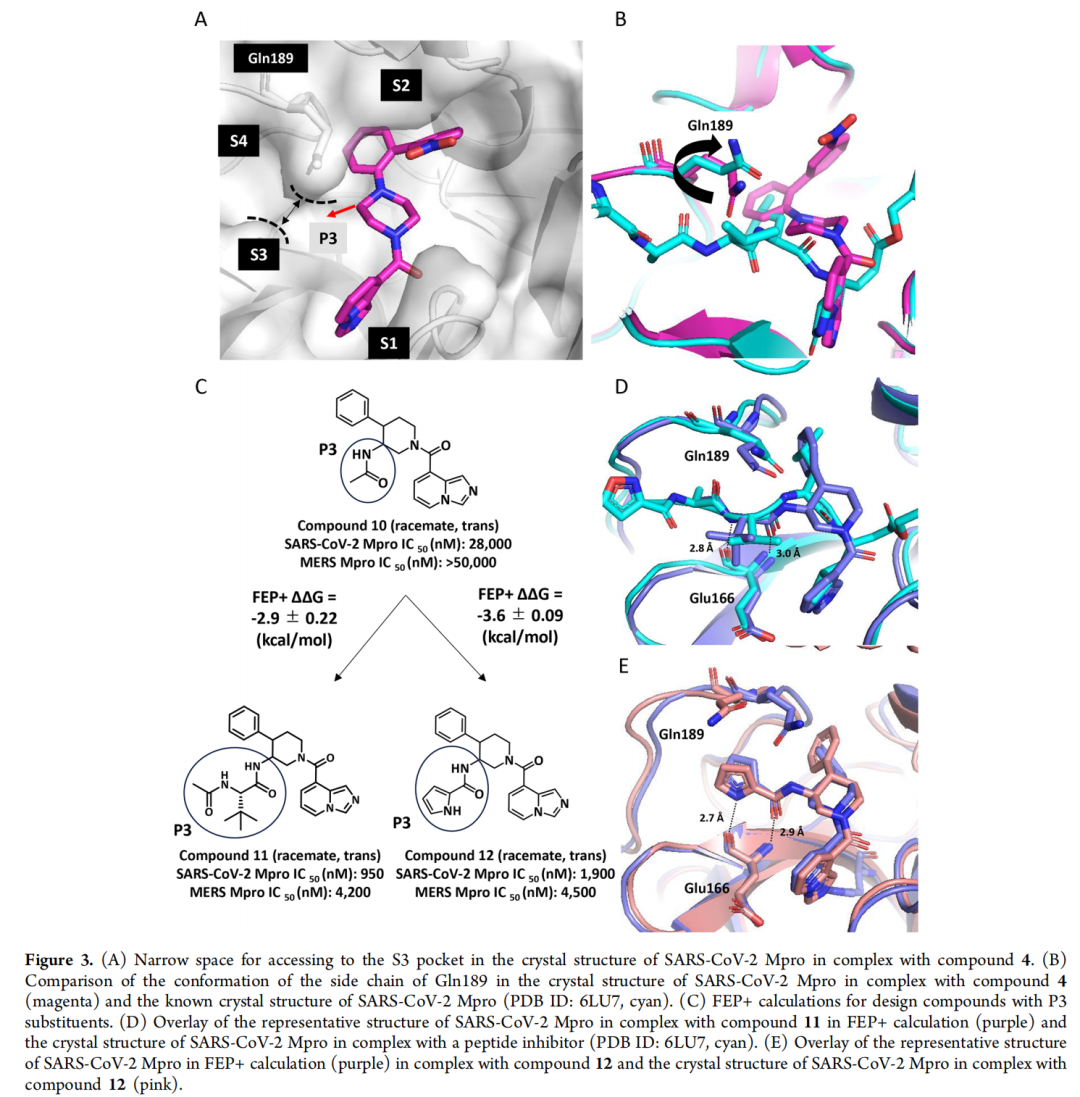
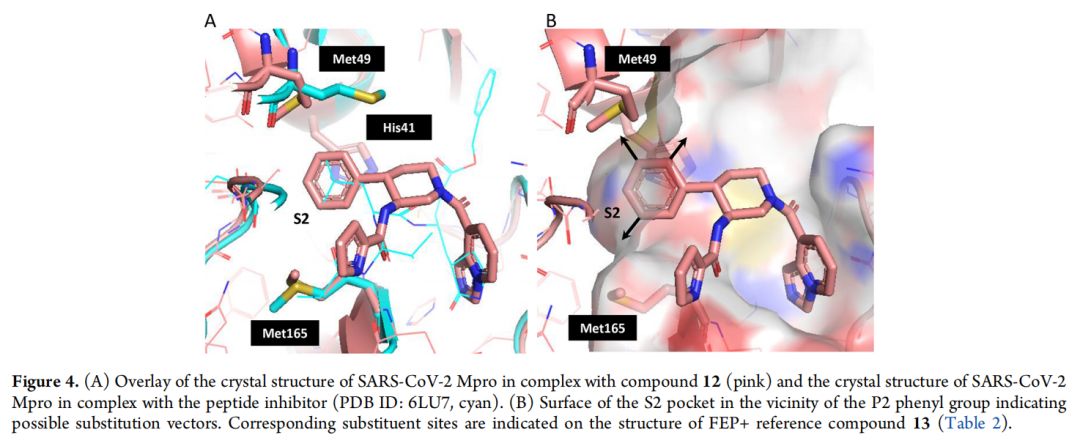
- S2口袋优化:针对S2口袋的疏水特性(由Met49、Met165组成),通过FEP筛选发现引入2,4-二氟苯基可增强范德华作用,使活性进一步提升至17-21 nM。
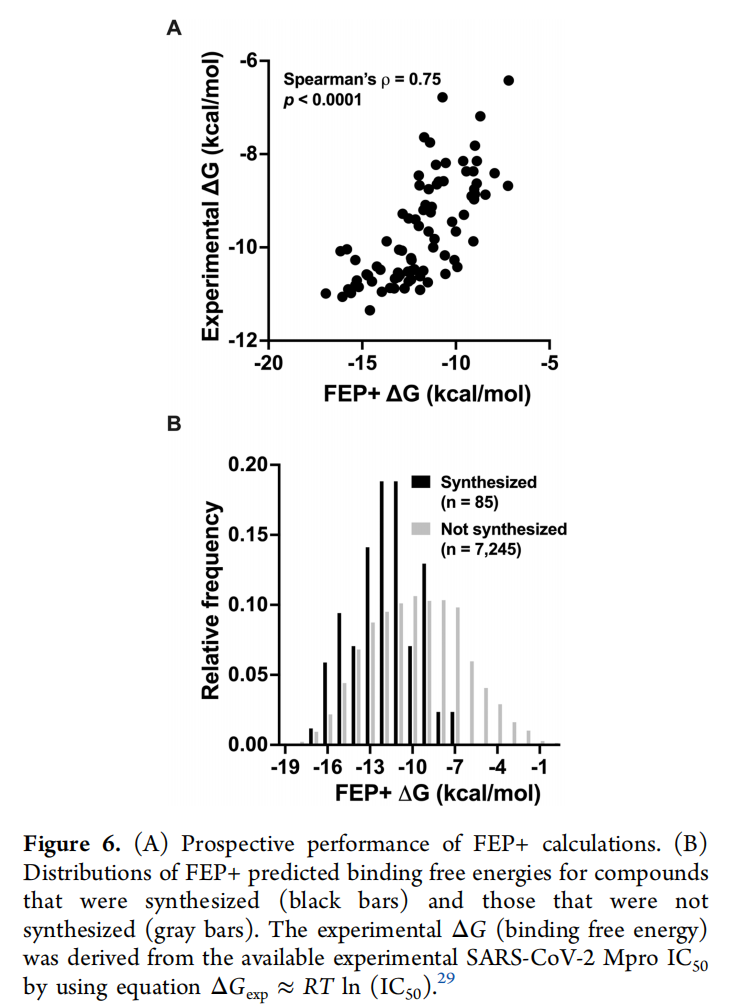
更重要的是,FEP计算展现出极高的预测准确性:7330个虚拟化合物中,85个合成化合物的预测结合能与实验IC₅₀值的Spearman相关系数达0.75(p<0.0001),这意味着CADD技术已能实现 设计即有效 的研发效率。
4. 成药性优化:兼顾活性与临床适用性
高活性化合物需具备良好的药代动力学特性才能进入临床,研究团队针对前期化合物代谢稳定性差的问题(人肝微粒体清除率CLint=85 mL/min/kg),通过降低脂溶性和引入羟基增强水溶性,最终得到化合物30。其成药性数据表现优异:
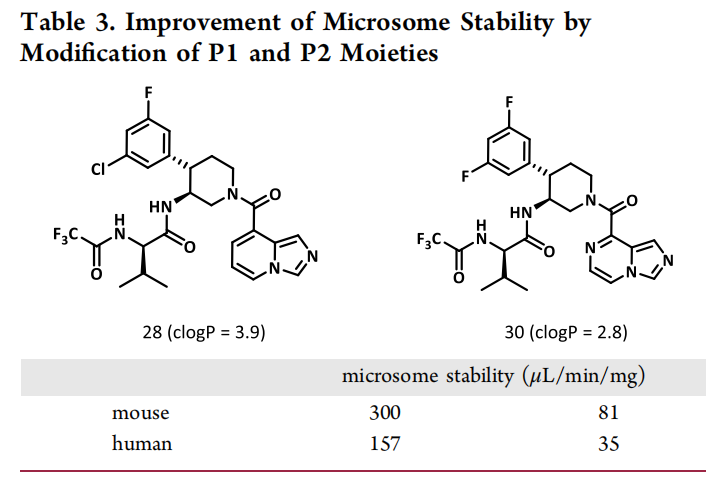
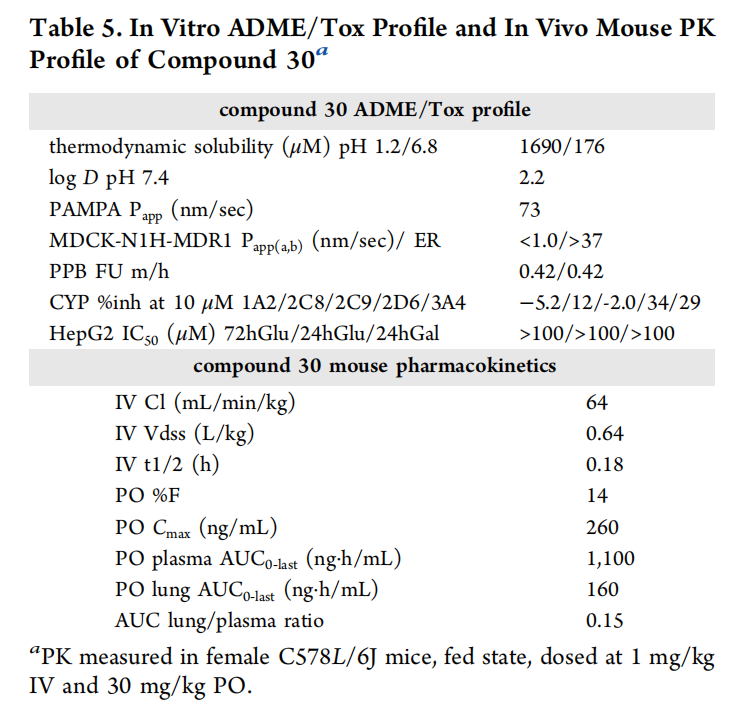
四、化合物30的性能突破:与现有药物的多维度对比
化合物30的核心价值通过与奈玛特韦、恩赛特韦的头对头比较得以凸显,其在活性、广谱性、安全性等关键指标上实现全面超越。

五、研究的科学价值与未来挑战
1. 学术与技术层面的双重贡献
- 构建广谱非共价抑制剂新骨架:验证了三取代哌啶母核作为Mpro非共价抑制剂的可行性,为后续衍生物开发提供了结构模板;
- 确立CADD研发新范式:从虚拟筛选到FEP优化的全流程技术体系,将"Hit到Lead"的缩短研发周期,较传统方法效率提升显著,为抗病毒药物快速研发提供了可复制的技术路线;
- 深化Mpro抑制机制认知:通过高分辨率共晶结构揭示了非共价抑制剂与S3、S2口袋的作用细节,为设计更高活性化合物提供了分子层面的理论依据。
2. 产业化进程中的关键挑战
尽管化合物30展现出巨大潜力,但其走向临床仍需突破两大瓶颈:一是口服生物利用度有待提升,需通过制剂优化(如纳米粒包合)或结构修饰(引入促吸收基团)进一步提高;二是缺乏体内药效学数据,需在K18-hACE2小鼠等动物模型中验证其降低病毒载量、改善肺部病理损伤的效果。此外,长期安全性评估(如重复给药毒性试验)也是进入临床试验前的必要环节。
总结
这篇研究通过精准的理性设计突破了现有抗新冠药物的多重局限,其开发的化合物30以"非共价结合、广谱活性、低药物相互作用"的核心优势,成为抗冠状病毒药物研发的重要成果。更重要的是,该研究充分展现了计算机辅助药物设计在加速创新药物研发中的核心价值,为应对未来病毒变异和新发突发传染病提供了"精准、高效、广谱"的药物研发新思路。随着后续体内研究和临床转化的推进,这类新型抑制剂有望成为下一代抗冠状病毒治疗的核心药物。
参考文献:Atsutoshi Okabe, Daniel W. Carney, Michiko Tawada, Thamina Akther, Jumpei Aida, Terufumi Takagi, Douglas R. Dougan, Abba E. Leffler, Jeffrey A. Bell, Leah Frye, Eugene R. Hickey, Mallareddy Komandla, Will Tao, Jangir Selimkhanov, Kazuko Yonemori, Edcon Chang, Kumar Saikatendu, and Atsuko Ochida, Discovery of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 Main Protease through Computer-Aided Drug Design, Journal of Medicinal Chemistry, 2025.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-10-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号