没有生物学重复的转录组差异分析如何挑选基因呢: 变化倍数与P值选谁?

没有生物学重复的转录组差异分析如何挑选基因呢: 变化倍数与P值选谁?

生信技能树
发布于 2024-12-27 19:47:37
发布于 2024-12-27 19:47:37
我们的生信入门课程遇到一个学员提问,他的提问非常清晰,有前因后果:
GSE编号:GSE144169 实验方法:高通量测序法 物种:智人 问题: 我想利用这个数据集寻找哪些基因是上调的,哪些基因是下调的,所以我做了如下尝试 1.首先我用的是小洁老师用的转录组代码,但是这里面只有两个样本,所以采用小洁老师上课的代码行不通
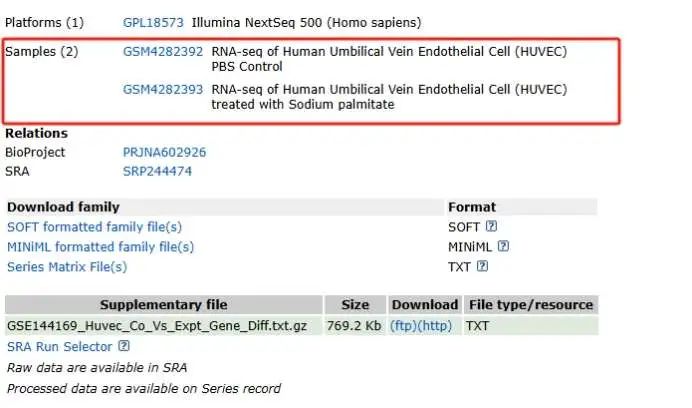
- 后来我看附加文件中,已经将差异基因写成文本格式,发在附加文件下,我就直接下载了,将文件导入,输入列名,发现没有p.value值,但是有log2FC值,所以我想问一下能不能用Huvec_Co和Huvec_Expt计算出p.value。
这个是差异基因的截图
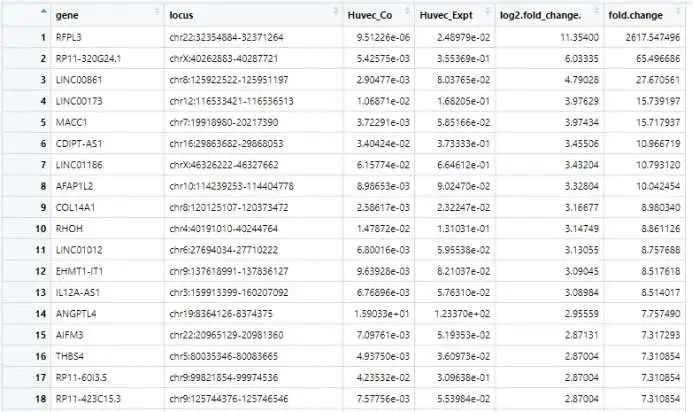
这个是列名的截图
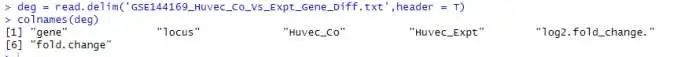
这个问题里面涉及到两个问题:
1、没有生物学重复的时候 可以使用 FC 值 即倍数变化 筛选差异基因吗?
2、没有生物学重复的时候 还有算法可以做差异分析吗?进而得到一个统计学显著性Pvalue值。
先看第一个:
毫无疑问,FC值 是基因在两组样本或者这里的一对一样本中的倍数变化值,在早期生物信息分析里面筛选差异基因的时候,常用的指标就是这个FC值,是可以用来筛选差异基因的,如使用阈值:FC > 2 或者FC < 1/2。
但是FC值有一个比较大的缺点,就是容易受到较小数值的影响(部分基因):
如: genei 在 A 组表达均值为 0.1,在 B 组中表达均值为 0.5,他们的差值只有 0.4,是非常微小的,但是 FC 达到了 5 倍 又: genei 在 A 组表达均值为 2,在 B 组中表达均值为 5,他们的差值达到了 3,但是 FC 只有 2.5 倍
绘图看一下基因表达均值与FC值的散点图:
all <- read.table("GSE144169_Huvec_Co_Vs_Expt_Gene_Diff.txt.gz",header=T,sep="\t")
head(all)
all$mean <- rowMeans(all[,c(3,4)])
qplot(log10(all$mean), all$log2.fold_change., ylab="Log2FC", xlab="Log10(Exp Mean)", size=I(0.7))
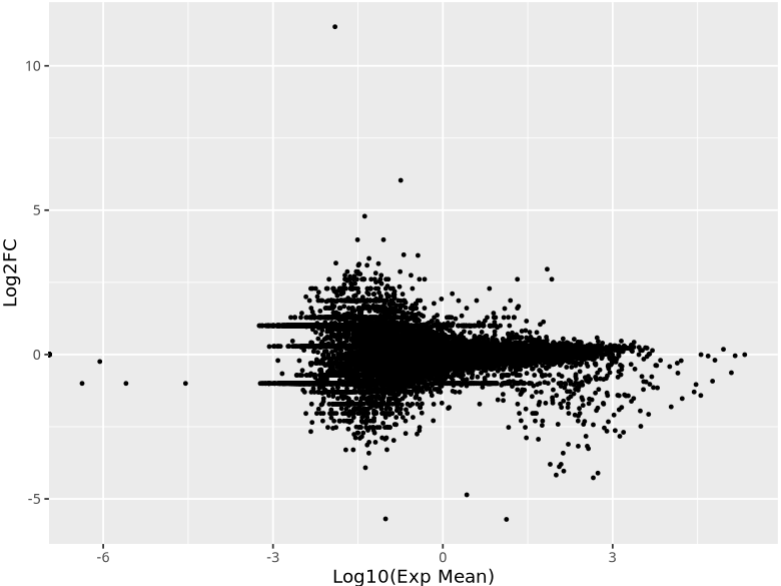
且没有一个统计学指标来判断这个基因的倍数变化是不是具有统计学意义。

上面这个GEO数据:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144169,提供的结果 是可以使用FC来进行差异基因筛选的。
对于第二个问题
当然,还是有算法可以对 这种只有一个 样本分组的差异进行分析,如转录组差异分析三大算法之一的edgeR,分析代码如下:
首先下载count矩阵:
# 加载R包
library(data.table)
library(tinyarray)
# load counts table from GEO
#urld <- "https://www.ncbi.nlm.nih.gov/geo/download/?type=rnaseq_counts&acc=GSE224401&format=file&file=GSE224401_raw_counts_GRCh38.p13_NCBI.tsv.gz"
acc <- "GSE144169"
urld <- "https://www.ncbi.nlm.nih.gov/geo/download/?type=rnaseq_counts&"
path <- paste0(urld, "acc=", acc, "&format=file&file=",acc,"_raw_counts_GRCh38.p13_NCBI.tsv.gz");
file <- paste0(acc,"_raw_counts_GRCh38.p13_NCBI.tsv.gz")
path
file
download.file(url = path, destfile = file)
tbl <- as.matrix(data.table::fread(file, header=T, colClasses="integer"), rownames=1)
dim(tbl)
ensembl_matrix=as.data.frame(tbl)
library(AnnoProbe)
library(org.Hs.eg.db)
keytypes(org.Hs.eg.db)
e2s<-AnnotationDbi::select(org.Hs.eg.db,
keys= rownames(ensembl_matrix),
columns="SYMBOL",
keytype = "ENTREZID")
head(e2s)
ids = na.omit(e2s)
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENTREZID),]
head(ids)
symbol_matrix= ensembl_matrix[match(ids$ENTREZID,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
symbol_matrix[1:4,]
接着拿到样本表型信息:
library(AnnoProbe)
library(GEOquery)
acc
gset = getGEO(acc, destdir = '.', getGPL = F)
pd = pData(gset[[1]])
pd[,1:5]
colnames(pd)
rownames(pd)
colnames(symbol_matrix)
com <- intersect(rownames(pd), colnames(symbol_matrix))
symbol_matrix <- symbol_matrix[, com]
pd=pd[com,]
pd$title
group_list=ifelse(grepl('Control',pd$title ), 'control','case' )
# 保存数据
group_list
table(group_list)
group_list = factor(group_list,levels = c('control','case' ))
dat=log10(edgeR::cpm(symbol_matrix)+1)
save(symbol_matrix,dat,group_list,file = 'step1-output.Rdata')
使用edgeR做一对一的差异分析:
rm(list=ls())
library(edgeR)
load("step1-output.Rdata")
ls()
# filter low expression gene
count_data <- symbol_matrix
keep <- rowSums(count_data)>0
filter_count <- count_data[keep,]
############## begin deg analysis
y <- DGEList(counts=filter_count, group=group_list)
y <- calcNormFactors(y)
dis <- 0.16 # for human
et <- exactTest(y, dispersion=dis)
res <- as.data.frame(topTags(et,dim(et)[1]))
head(res)
结果如下:
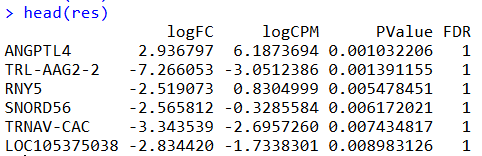
可以比较一下edgeR算法的logFC与这个数据集给出的FC值
## compare
head(res)
head(all)
com_gene <- intersect(rownames(res), all$gene)
dat_plot <- data.frame(com_gene,as.numeric(res[com_gene, "logFC"]), as.numeric(all[match(com_gene,all$gene), 5]))
head(dat_plot)
colnames(dat_plot) <- c("gene", "logFC", "edgeR_logFC")
head(dat_plot)
library(ggpubr)
p=ggscatter(dat_plot, x = "logFC", y = "edgeR_logFC",
color = "black", shape = 21, size = 2, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.sep = "\n")
)
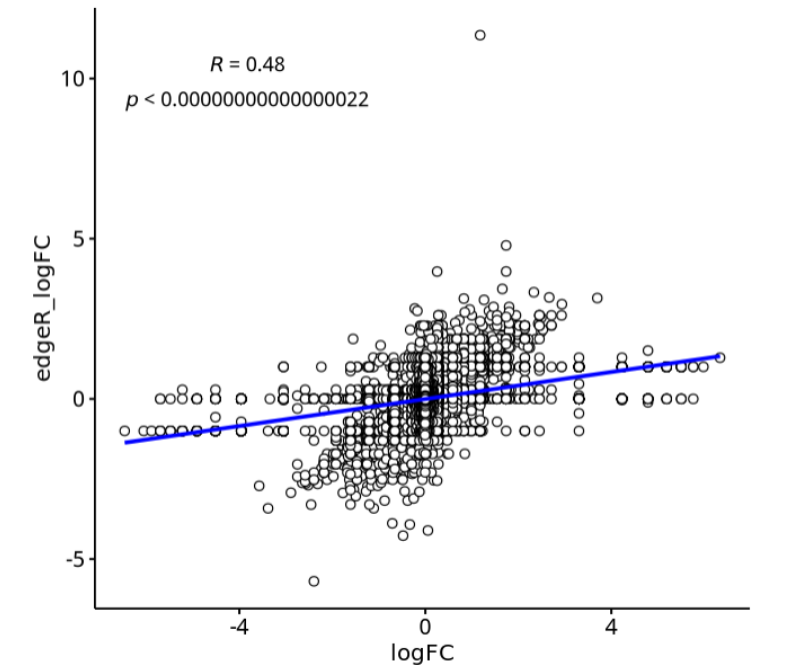
p
结果如下:
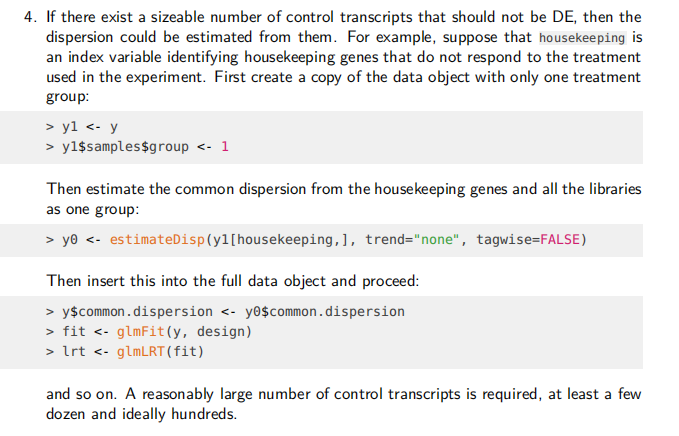
edgeR 算法给出的建议:What to do if you have no replicates

他们公出了四点建议:但是任何一点都不是可以替代 有生物学重复的好方案 (千万要有组内重复样品设计)
第一条也是最好的一条,直接使用FC值来筛选基因进行后续的研究
不要试图去找一个统计学显著性。
- Be satisfied with a descriptive analysis, that might include an MDS plot and an analysis of fold changes. Do not attempt a significance analysis. This may be the best advice
第二条是我上面的代码,给了一个 dispersion值
- Simply pick a reasonable dispersion value, based on your experience with similar data, and use that for exactTest or glmFit. Typical values for the common BCV (square-root dispersion) for datasets arising from well-controlled experiments are 0.4 for human data,0.1 for data on genetically identical model organisms or 0.01 for technical replicates
第三条:

第四条:
FC值与Pvalue如何选择,你现在有答案了吗?
可参考:FC值 结合 过滤基因表达值指标如基因在两个样本中的均值以及差值。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-12-25,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号