Nat Comput Sci | 给羊驼一套化学语言,它开始学会设计分子

Nat Comput Sci | 给羊驼一套化学语言,它开始学会设计分子

MindDance
发布于 2026-05-15 14:14:00
发布于 2026-05-15 14:14:00
一分钟速读
这篇发表于 Nature Computational Science 的研究,由 Joseph M. Cavanagh 等与 Teresa Head-Gordon 团队完成,作者主要来自加州大学伯克利等机构。它讨论的不是让大语言模型回答几个化学问题,也不是让模型给药物研发人员当聊天助手,而是把一个开源权重大模型 Meta-Llama-3.1-8B-Instruct 改造成能够直接输出药物分子字符串的 化学语言模型。作者将这个模型命名为 SmileyLlama。
它的训练思路很直接:用约 200 万个来自 ChEMBL v33 的药物样分子,把分子性质和分子结构配成训练样本。输入端是用户想要的性质,例如分子量、logP、氢键供体/受体、拓扑极性表面积、是否含有特定子结构;输出端是对应的 SMILES 分子字符串。随后,作者再用 直接偏好优化 让模型更听指令,并把它接入 iMiner 框架,用 SARS-CoV-2 主蛋白酶作为案例,测试模型能否在三维蛋白口袋约束下生成候选抑制剂分子。
结果显示,SmileyLlama 在 GuacaMol 分子生成基准上达到了较强表现:有效性 0.958、唯一性 1.000、新颖性 0.987。相比之下,原始 Llama 在零样本条件下虽然能写出一部分合法 SMILES,但唯一性只有 0.457;给 20 个示例后,唯一性上去了,有效性却降到 0.465。换句话说,通用大模型知道一点化学语法,并不等于它会稳定地生成可用分子。
更重要的是,这篇文章把一个通用开源大模型,从会谈论化学,推进到能够按药物化学条件直接生成分子。监督微调让模型学会在属性范围内生成分子,直接偏好优化进一步提高了模型遵守约束的能力。不过边界也清楚:大环化合物、精确数值约束、物理上不合理的请求仍然困难;分子对接得分也只是计算筛选信号,不等于真实药效。
研究背景:为什么分子生成不能只靠会聊天的大模型
药物发现里有一个长期难题:我们真正想找的,不是一串漂亮的字符,而是一个能够被合成、能进入体内、能到达靶点、能与蛋白口袋结合、还尽量少产生毒副作用的分子。这个要求听起来像一句话,落到计算上却是多重约束的组合:分子是否合法、是否新颖、是否具有类药性、是否避开不稳定结构、是否有合理的分子量和脂水分配系数、是否能在三维空间里贴合靶点活性口袋。
过去几年,化学语言模型已经证明了一件事:分子也可以被看成一种语言。SMILES 把分子图压缩成一串字符,SELFIES 则进一步强调字符串到分子的鲁棒映射。只要模型学会这种字符序列的统计规律,就可以像写句子一样写分子。于是,研究者用 ChEMBL、ZINC 等大规模分子数据库训练了很多从头分子生成模型,包括变分自编码器、循环神经网络、GPT 类模型,以及结构化状态空间序列模型。
但药物设计的问题从来不只是会不会写分子。一个模型随机生成一批合法分子,并不等于它能按药物化学家的意图工作。药物化学家通常不会只说生成一个分子,而是会给出一组很具体、但又不完全精确的要求:分子量不要太大,logP 不要太高,氢键供体别太多,拓扑极性表面积落在某个范围,保留某个片段,避开某类反应性基团,或者在蛋白口袋里尽量形成更好的结合。
这就是传统化学语言模型和通用大语言模型各自的短板。前者擅长分子字符串,却不天然理解自然语言指令;后者擅长自然语言,却不能保证输出的是合法、互异、可用于化学计算的 SMILES。很多人把大模型用于化学时,首先想到的是让它查资料、解释反应、辅助写实验记录,甚至作为虚拟实验室成员。SmileyLlama 走的是另一条路:不是让大模型谈论化学,而是让大模型直接用化学语言说话。
Lipinski 五规则、Veber 规则、拓扑极性表面积、可旋转键数量等指标,本质上不是药物成功的定律,而是早期筛选中的经验尺子。它们帮助研究者排除明显不合适的分子,但不会自动给出真正有效的药物。一个模型如果只优化对接得分,往往容易偏向更大、更疏水、相互作用更多的分子;这些分子在计算口袋里看起来得分高,现实中却可能溶解性差、代谢差、合成困难。因此,一个实用的生成模型,必须在 探索化学空间 和 遵守药物化学约束 之间找到平衡。
SmileyLlama 的意义就在这里:它尝试把大语言模型的指令理解能力、化学语言模型的分子生成能力,以及强化学习式的目标优化能力放进同一套流程里。这个问题并不华丽,但很关键。因为药物设计的真实场景通常不是单目标优化,而是一堆约束同时压过来。
研究设计:把 Llama 改造成 SmileyLlama
作者选用的基础模型是 Meta-Llama-3.1-8B-Instruct。选择开源权重大模型有现实优势:研究者可以自己微调权重或适配器,不必把潜在有价值的分子数据传到远程闭源接口;也可以控制推理参数、训练算法,并做后续可解释性分析。
SmileyLlama 的训练分为两步。
第一步是 监督微调。作者从 ChEMBL v33 取约 200 万个分子,用 RDKit 计算药物化学相关性质,包括分子量、logP、氢键供体和受体数量、拓扑极性表面积、sp3 碳比例、可旋转键、大环结构、不良 SMARTS 子结构、共价弹头相关 SMARTS 模式、指定子结构以及分子式等。每个分子都会被包装成一个训练样本:输入是带有若干属性条件的提示,输出是满足这些属性的 SMILES。
这里有个细节很值得注意:训练时,每个属性有 50% 的概率被写入提示。这不是随手设计。药物研发中的真实需求往往是不完整的,有时只关心分子量和 logP,有时还要控制氢键数量,有时只想保留某个片段。把属性随机写入提示,相当于让模型学会在不同信息量下工作,而不是只适应一种固定模板。
第二步是 直接偏好优化。作者先让监督微调后的模型按某个性质要求生成多个 SMILES,再用 RDKit 判断它们是否符合要求。符合要求的分子被当作优选答案,不符合要求的分子被当作劣选答案,形成成对偏好数据。DPO 的作用就是更新模型,使它更倾向于生成优选分子,而不是劣选分子。相比传统强化学习,这种做法不需要额外训练一个奖励模型,工程上更轻。
这套方法后来也被嵌入 iMiner。原始 iMiner 用循环神经网络生成分子,再用 AutoDock Vina 进行分子对接,把对接得分和类药性得分反馈给模型。本文用 SmileyLlama 替换掉原先的分子生成器,并用 DPO 替换更重的 PPO 优化流程。这样,模型既可以通过自然语言提示接收药物化学约束,也可以通过蛋白三维结构筛选得到目标相关的偏好信号。
这不是简单换了一个更大的模型。真正的改动是:把分子设计问题重新表述成一类可被大语言模型学习的条件生成任务。
实验结果:从会写合法分子,到按目标探索化学空间
1. 基本生成能力:通用 Llama 会一点 SMILES,但不稳定
作者先用 GuacaMol 基准测试 SmileyLlama 的基础分子生成能力。这个基准主要看几类指标:生成字符串是否能被 RDKit 解析为合法分子,生成分子是否互异,是否不在训练集中,以及生成分布是否接近训练数据的药物样分子分布。
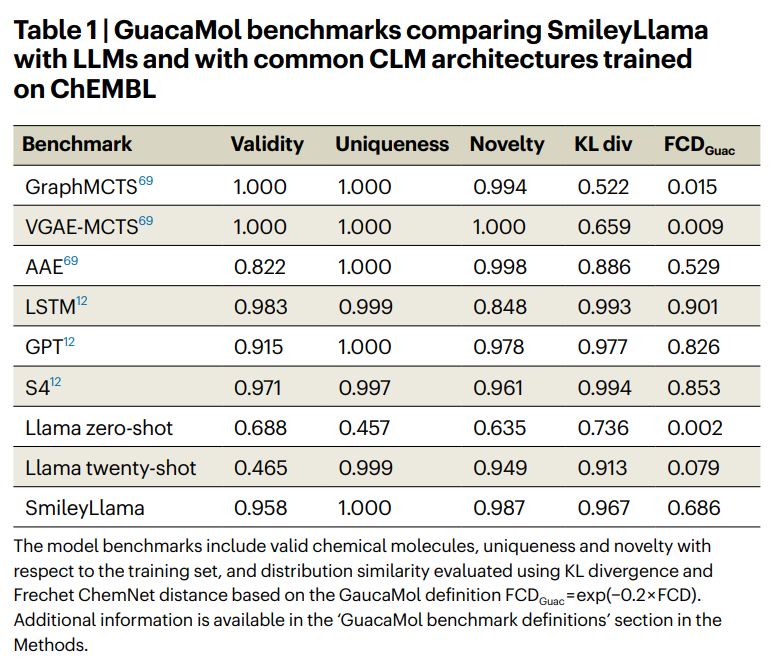
不同模型在 GuacaMol 分子生成基准上的表现。Validity、Uniqueness、Novelty 分别表示合法性、唯一性和新颖性;KL div 与 FCDGuac 用于衡量生成分布与 ChEMBL 训练分布的接近程度。
不同模型在 GuacaMol 分子生成基准上的表现。Validity、Uniqueness、Novelty 分别表示合法性、唯一性和新颖性;KL div 与 FCDGuac 用于衡量生成分布与 ChEMBL 训练分布的接近程度。
上表里最直观的对比,是原始 Llama 和 SmileyLlama 的差距。零样本 Llama 的有效性为 0.688,说明它确实在预训练中接触过一些 SMILES 语法,能写出一部分合法分子;但唯一性只有 0.457,暗示它会重复某些熟悉结构,缺乏稳定的化学空间泛化能力。给它 20 个 ChEMBL 示例后,唯一性提高到 0.999,但有效性反而降到 0.465。这个结果很有画面感:模型被示例推着往外走,却没有真正掌握 SMILES 可以如何变形。
SmileyLlama 的表现明显不同。它的有效性达到 0.958,唯一性 1.000,新颖性 0.987,KL div 为 0.967,FCDGuac 为 0.686。这个结果说明,监督微调之后,模型不仅能写出合法分子,而且生成分布没有明显跑偏。需要补一句:SmileyLlama 并不是在所有分布指标上都超过传统从零训练的化学语言模型,例如 LSTM、GPT、S4 在部分分布相似性指标上仍然更高。更稳妥的判断是:它把一个通用指令模型拉到了现代化学语言模型可比较的区间,同时获得了自然语言条件控制的入口。
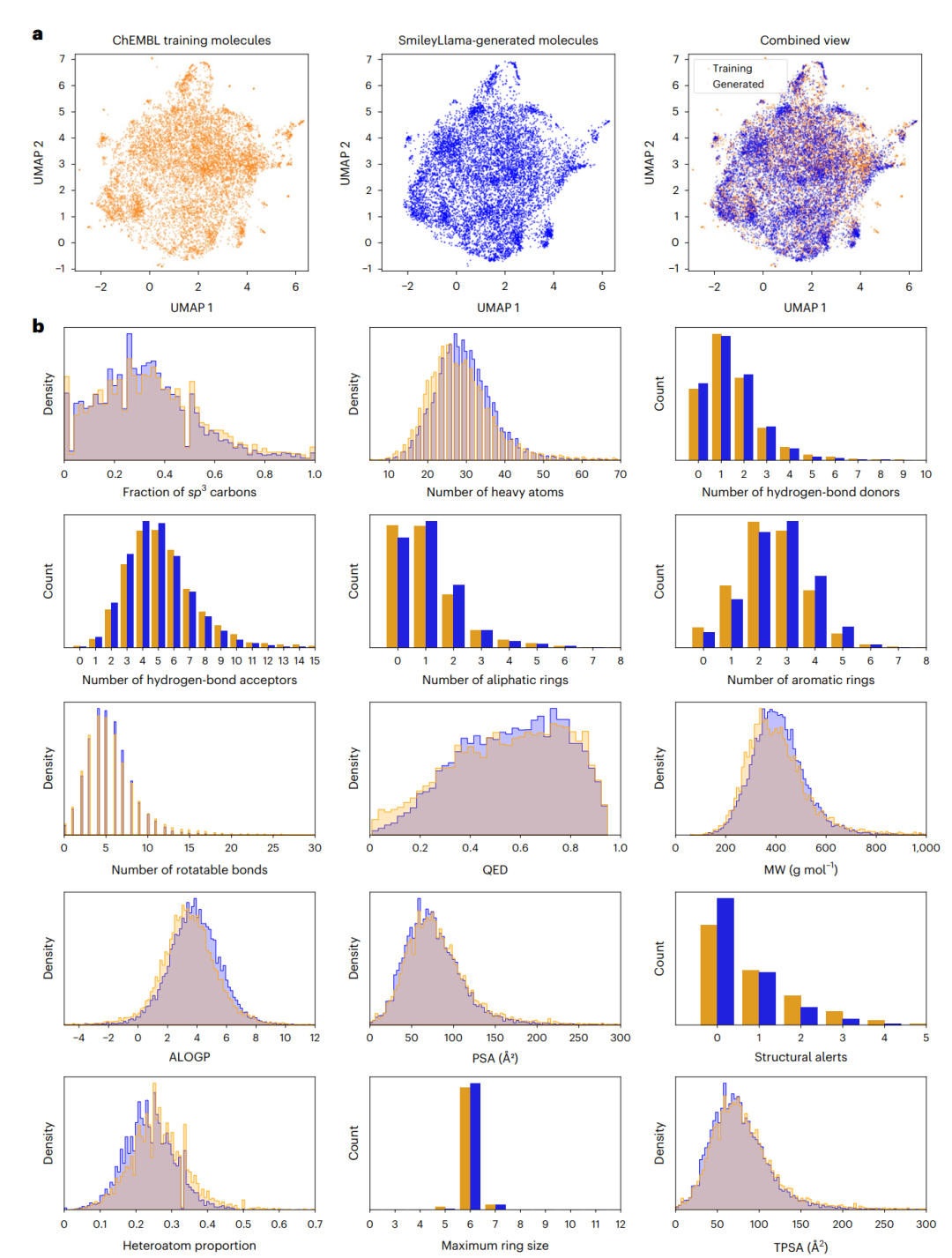
SmileyLlama 生成分子与 ChEMBL 训练分子的化学空间和药物化学属性分布比较。蓝色为生成分子,金色为训练分子;上方为 UMAP 二维投影,下方为多项分子性质分布。
SmileyLlama 生成分子与 ChEMBL 训练分子的化学空间和药物化学属性分布比较。蓝色为生成分子,金色为训练分子;上方为 UMAP 二维投影,下方为多项分子性质分布。
上图可以先看 UMAP。生成分子与训练分子在二维化学空间中大范围重叠,说明模型没有只在局部小区域里打转。再看下面的性质分布,sp3 碳比例、重原子数量、氢键供体和受体、芳香环与脂肪环数量、可旋转键、QED、分子量、ALOGP、PSA、TPSA 等指标都与 ChEMBL 有较好对应。对药物生成模型来说,这比单纯有效率更重要。因为一个模型完全可以生成大量合法但奇怪的分子,指标看起来漂亮,真正用于筛选时却没有价值。
2. 属性控制:范围约束做得好,稀有结构和精确数值仍难
药物化学里,很多约束不是精确值,而是范围。例如分子量不超过 500,logP 不超过 5,氢键供体不超过 5,氢键受体不超过 10。作者专门设计了一组属性控制测试,每项任务生成 1000 个分子,然后统计其中同时满足 有效、互异、符合指定性质 的比例。
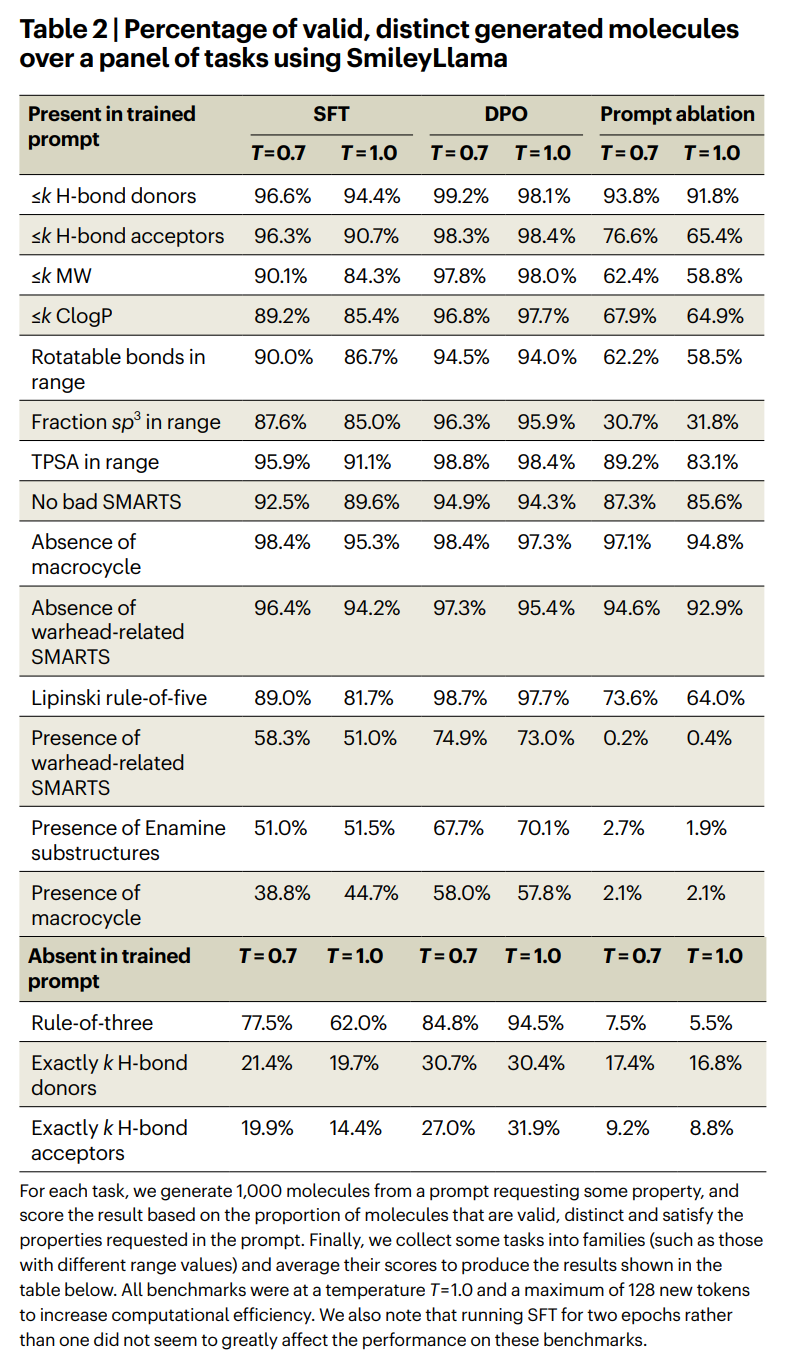
SmileyLlama 在不同属性约束任务中的生成成功率。每项任务生成 1000 个分子,统计有效、互异且符合约束的分子比例。DPO 在多数范围约束上提升明显,但对大环、精确数值和稀有结构仍然困难。
SmileyLlama 在不同属性约束任务中的生成成功率。每项任务生成 1000 个分子,统计有效、互异且符合约束的分子比例。DPO 在多数范围约束上提升明显,但对大环、精确数值和稀有结构仍然困难。
上表传递的信息很清楚。只经过监督微调时,SmileyLlama 已经能较好处理常见范围约束。例如在温度为 1.0 时,氢键供体上限任务达到 94.4%,氢键受体上限任务达到 90.7%,拓扑极性表面积范围任务达到 91.1%,Lipinski 五规则任务达到 81.7%。
经过 DPO 后,模型的遵守指令能力明显提高。仍以温度 1.0 为例,分子量上限任务从 84.3% 提升到 98.0%,logP 上限任务从 85.4% 提升到 97.7%,Lipinski 五规则从 81.7% 提升到 97.7%。这说明 DPO 对这类可自动判定的药物化学约束非常有效。
但弱点也被表格直接摆出来。要求模型生成含有大环的分子时,监督微调在温度 1.0 下只有 44.7%,DPO 后也只有 57.8%。要求含有共价弹头相关 SMARTS 模式,DPO 后为 73.0%;要求含有 Enamine 子结构,DPO 后为 70.1%。这些任务更依赖数据中少见结构的覆盖,也更考验模型对局部子结构的精确控制。
精确数值任务更明显。训练时作者主要使用范围约束,而不是精确指定某个氢键供体或受体数量。因此,当任务变成精确 k 个氢键供体或精确 k 个氢键受体时,即使 DPO 后也只有约 30% 左右。这个结果反过来提醒我们:模型学会的是训练任务定义中的化学控制方式,而不是抽象地掌握所有化学计数规则。
还有一个关键对照:作者做了属性提示消融,也就是训练时去掉分子属性信息,只让模型学会从统一提示生成分子。这个消融模型在常见通用任务上还能勉强工作,但一碰到大环、共价弹头、Enamine 子结构这类稀有或条件性任务,表现迅速塌下去。也就是说,SmileyLlama 的能力不是凭空来自 Llama 的预训练知识,而是来自 把药物化学属性认真写进训练提示 这件事。
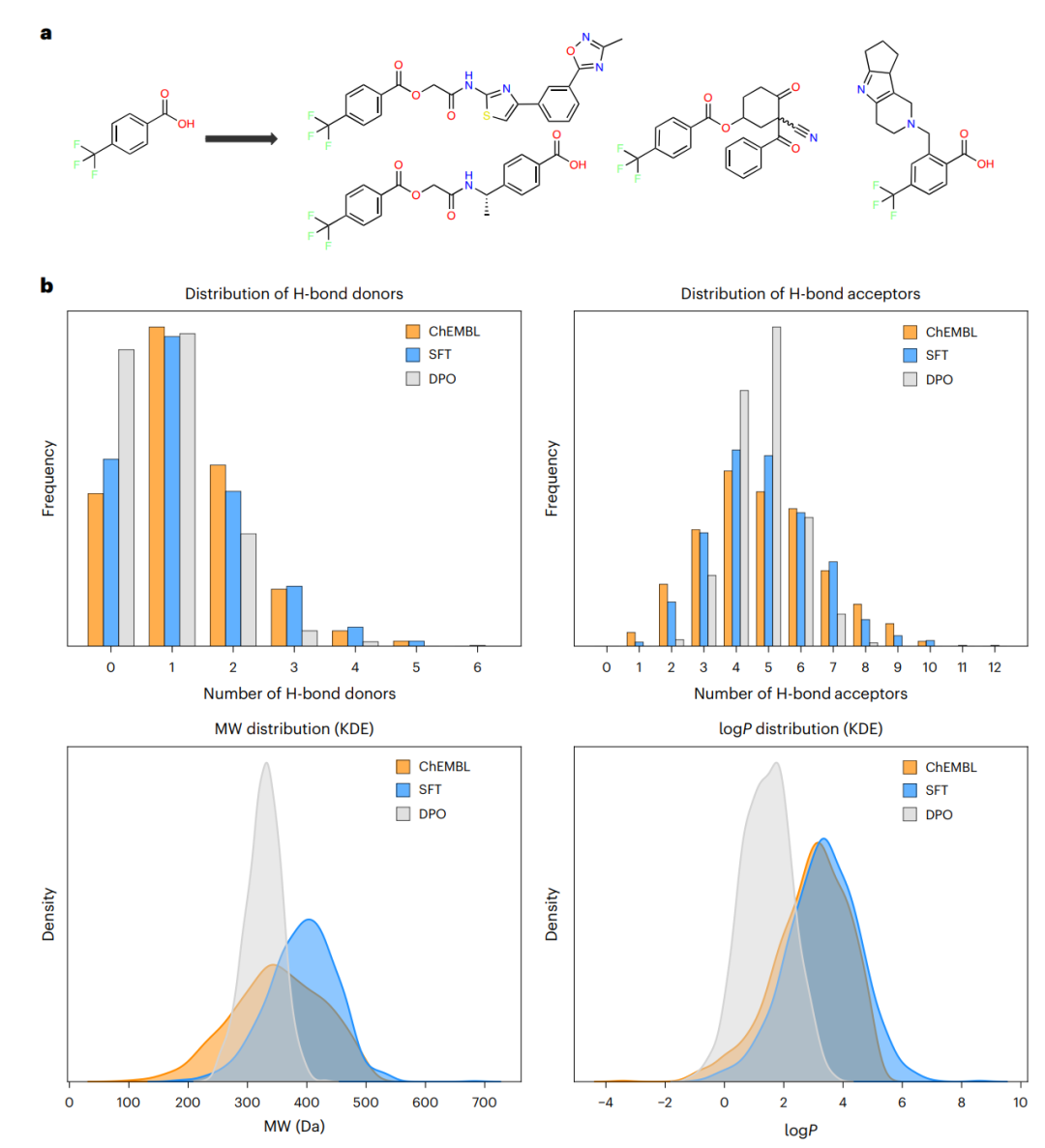
SmileyLlama 的条件生成示例与 DPO 前后分布变化。上方显示模型从指定片段出发生成符合 Lipinski 五规则的分子;下方比较 ChEMBL、监督微调模型和 DPO 模型在氢键供体、氢键受体、分子量和 logP 上的分布差异。
SmileyLlama 的条件生成示例与 DPO 前后分布变化。上方显示模型从指定片段出发生成符合 Lipinski 五规则的分子;下方比较 ChEMBL、监督微调模型和 DPO 模型在氢键供体、氢键受体、分子量和 logP 上的分布差异。
上图很好地展示了 DPO 的代价。灰色的 DPO 分布更集中,也更贴近用户设定的规则;蓝色的监督微调分布更宽,和 ChEMBL 过滤后的分布更相似。对应到真实研发流程,监督微调更像早期探索,适合在化学空间里保持多样性;DPO 更像后期收口,适合在明确约束下提高命中率。
这不是谁更好,而是用在不同阶段。早期如果过早把分布压窄,可能错过意外有价值的结构;后期如果一直保持太宽,筛选成本会很高。
3. 接入三维结构约束:以 SARS-CoV-2 主蛋白酶为案例
前面的实验主要关注二维分子结构和药物化学性质,但药物分子最终要进入蛋白口袋。一个候选分子是否值得继续看,至少要考虑它在三维空间里是否可能与靶点形成合理结合。
作者把 SmileyLlama 接入 iMiner,并用 SARS-CoV-2 主蛋白酶作为测试靶点。这个蛋白酶对病毒生命周期很关键,也有较多已解析三维结构,因此适合做结构基础分子设计的示范。模型每轮生成 2000 个有效 SMILES,通过 AutoDock Vina 进行对接,再结合 iMiner 的类药性评分构造优化信号。
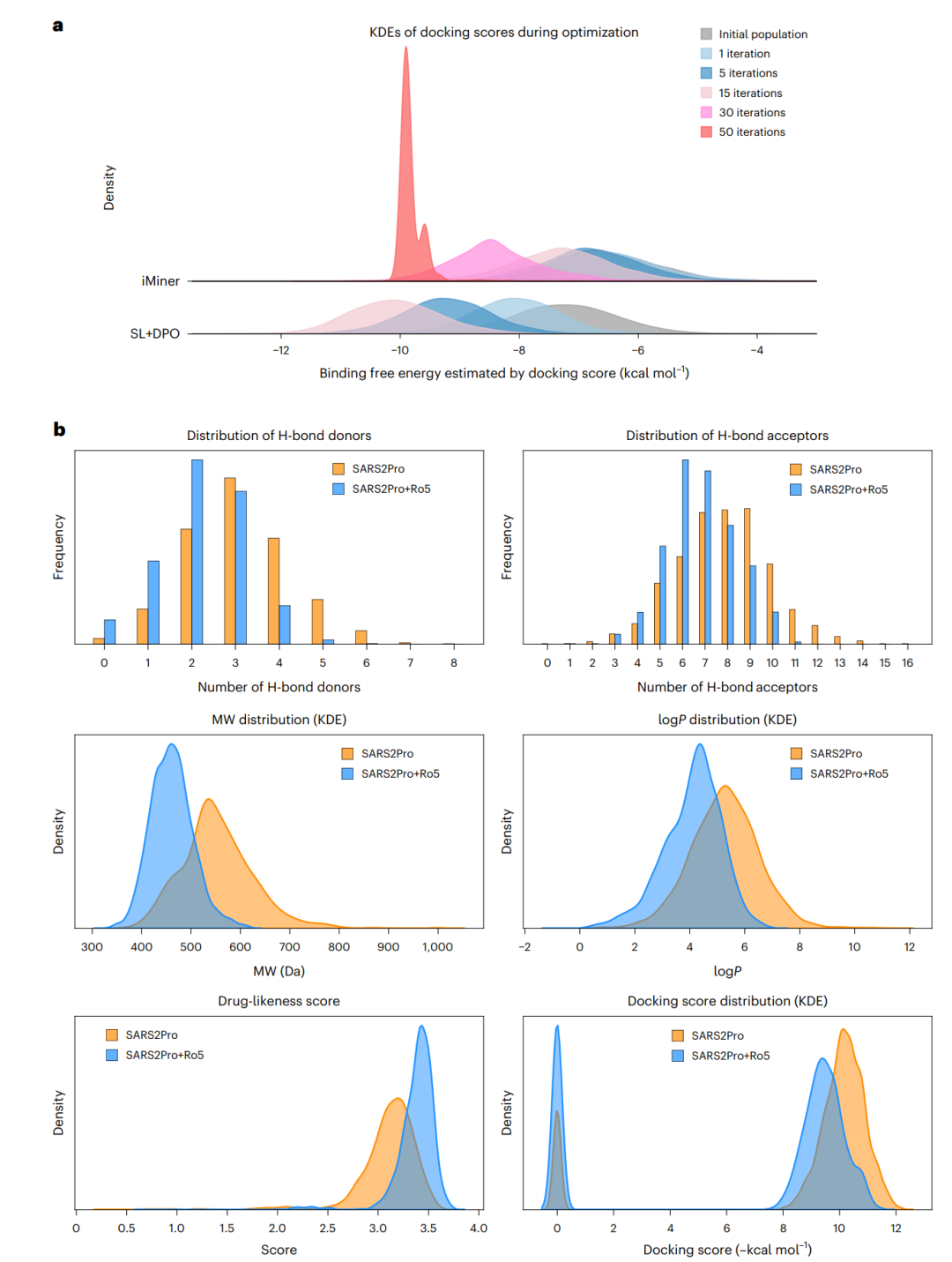
SmileyLlama/iMiner 针对 SARS-CoV-2 主蛋白酶的优化结果。上方为对接评分分布随优化迭代变化;下方比较只优化蛋白结合目标与叠加 Lipinski 五规则约束后的分子性质分布。
SmileyLlama/iMiner 针对 SARS-CoV-2 主蛋白酶的优化结果。上方为对接评分分布随优化迭代变化;下方比较只优化蛋白结合目标与叠加 Lipinski 五规则约束后的分子性质分布。
这组结果里有两个层次。
第一,SmileyLlama 比原始 iMiner 更高效。论文显示,SmileyLlama 大约只需要原始 iMiner 约四分之一的迭代轮数,就能达到相近的对接得分改善水平。原始 iMiner 在后期容易出现多样性坍缩,也就是越来越多生成分子集中到少数结构附近;SmileyLlama 则在提升对接得分的同时保持了更宽的分布。
第二,即便优化目标中已经包含 iMiner 的类药性评分,以对接表现为核心的优化仍可能带来药物化学偏差。只用 High SARS2PRO 目标时,生成分子的氢键供体和受体分布还可以,但分子量和 logP 不够理想。论文把这解释为原始 iMiner 奖励函数仍有不足:如果继续沿用传统化学语言模型流程,可能需要重新加权奖励函数、加入新项、调超参数,甚至重新训练模型。
SmileyLlama 的优势在这里显出来。作者把提示改成同时要求 High SARS2PRO 和 Lipinski 五规则相关限制,分子量、logP 和类药性评分明显改善,同时对接得分有所下降。这个下降并不意外,因为更小、更受约束的分子通常能形成的分子间相互作用更少。真正有价值的是:无需重新训练模型,只改提示,就能把生成分布往更药物样的方向推。
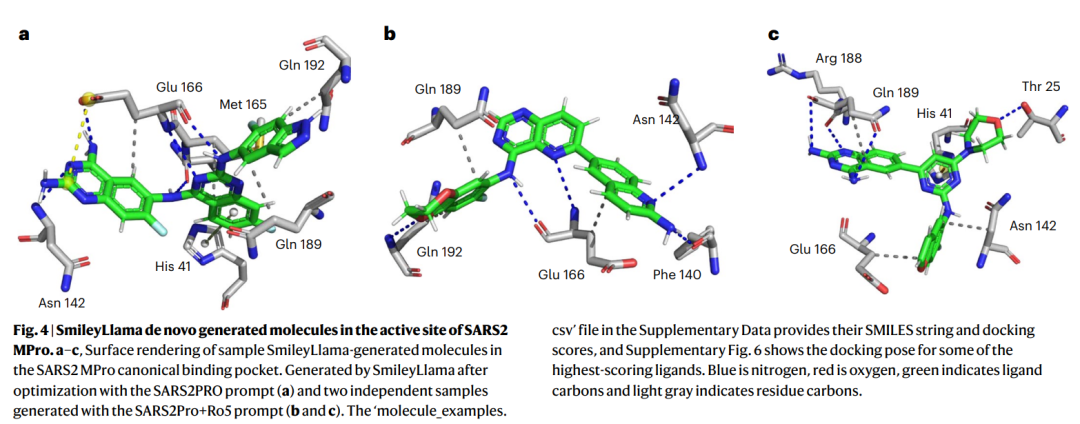
SmileyLlama 生成分子在 SARS-CoV-2 主蛋白酶活性口袋中的对接构象。绿色为配体碳原子,红色为氧,蓝色为氮,浅灰色为蛋白残基碳原子。
SmileyLlama 生成分子在 SARS-CoV-2 主蛋白酶活性口袋中的对接构象。绿色为配体碳原子,红色为氧,蓝色为氮,浅灰色为蛋白残基碳原子。
上图展示了生成分子在主蛋白酶经典结合口袋中的构象。作者还指出,部分高分分子与此前 Jorgensen 团队优化的 perampanel 三叶形抑制剂变体有相似之处;不同的是,Jorgensen 组分子保留中心吡啶酮环,而 SmileyLlama 生成的分子用嘧啶基团替换了这一三叶形核心。论文还报告,这些生成分子在 Therapeutic Target Database 中没有明显同源匹配,平均合成可及性评分约为 3,按该评分体系属于相对容易合成的候选结构。
这里要把话说准:这些结果说明模型可以提出有一定计算吸引力的候选结构,不能直接说明它发现了新冠治疗药物。对接分数、类药性评分、合成可及性评分都只是筛选信号,后续仍要经过合成路线评估、体外活性验证、选择性测试、ADMET、毒性、脱靶效应以及突变鲁棒性等步骤。SmileyLlama 解决的是候选生成和计算筛选的前端问题,不是药物发现的终点。
这项工作的真正价值:让大模型从化学助手变成化学生成器
很多大模型进入科学研究的方式,是作为一个会回答问题的助手。它可以解释名词、总结论文、写代码、规划实验流程。这些功能当然有用,但在分子设计里还不够。药物发现需要的是能产生结构、能被算法评分、能进入下一轮实验或计算流程的对象。
SmileyLlama 的路线更像是把大模型嵌入科研工作流:自然语言提示负责表达目标,化学字符串负责承载结构,RDKit 和分子对接负责提供外部检查,DPO 负责把检查结果反馈给模型。这种组合让大模型不只是站在化学旁边解释化学,而是进入化学空间本身。
它也给了领域模型训练一个现实启发。过去要做分子生成,通常会从零训练专门的化学语言模型。本文表明,一个通用开源权重大模型经过较轻量的监督微调和偏好优化,也能达到接近现代化学语言模型的生成表现。作者使用 LoRA 适配器和 4 张 A40 GPU 训练一个 epoch,论文中估算监督微调约 32 小时,成本约为 53 美元。这个数字不代表所有人都能复现同等成本,但至少说明,这条路径不是只有超大算力团队才能探索。
当然,SmileyLlama 也没有把问题全部解决。它对数据丰富、规则清晰、可自动判定的范围约束表现最好;对数据稀疏的大环结构、稀有共价弹头、精确计数和物理上矛盾的请求,仍然会出错。论文还展示了一个很典型的现象:当要求模型生成物理上不可能的分子条件时,它不会拒绝,而是生成一个不满足条件的 SMILES;在某些长输出场景下,模型还可能陷入重复模式。这些现象说明,SmileyLlama 还不是一个可靠的化学审稿人,它更像一个经过化学训练的候选分子生成器。
因此,读这篇文章最稳妥的方式,不是把它看成药物 AI 的终局答案,而是看成一个清晰的方向:用大语言模型承接人类目标,用化学工具约束模型输出,用偏好优化把反馈写回模型行为。这条链路一旦打通,未来可以扩展到合成规划、金属配合物设计、材料分子设计,甚至更复杂的生物分子工程。
总结讨论:化学空间开始有了新的交互方式
SmileyLlama 让人看到一种新的分子设计交互方式。过去,研究者往往需要写脚本、设奖励函数、调超参数,再从成千上万个生成分子里筛掉大部分不合适结构。现在,至少在这项研究展示的范围内,用户可以直接把约束写进提示,让模型生成更接近目标的分子,再通过 RDKit、对接和 DPO 进入下一轮筛选。
这不是把药物发现变成一句提示词,也不是把化学规律交给模型自由发挥。真正的关键在于,模型输出的每一步都被外部化学工具检查和反馈。大模型提供的是语言接口和生成能力,化学信息学工具提供的是规则约束,分子对接提供的是结构场景,偏好优化提供的是迭代方向。几者合在一起,才构成一个相对可信的候选分子探索流程。
这篇文章留下的余味是:当大模型开始直接写分子,化学空间不再只是数据库里的结构集合,也不只是算法在高维向量里的搜索区域。它开始变成一种可以被自然语言驱动、被实验规则约束、被计算反馈塑形的工作场景。模型能走多远,最终仍要看数据质量、目标定义、外部验证和真实实验。但从这项工作开始,让模型按研究者意图进入化学空间,已经不再只是一个概念演示。
参考文献
Cavanagh, J.M., Sun, K., Gritsevskiy, A. et al. SmileyLlama: modifying large language models for directed chemical space exploration. Nat Comput Sci (2026). https://doi.org/10.1038/s43588-026-00986-y
代码链接
http://github.com/THGLab/SmileyLlama
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号