Nature | STARLING实现无序蛋白构象集合的高精度预测

Nature | STARLING实现无序蛋白构象集合的高精度预测

DrugOne
发布于 2026-03-02 19:08:28
发布于 2026-03-02 19:08:28
DRUGONE
内在无序蛋白及其区域在几乎所有真核生物细胞过程中都发挥关键作用,但它们并不存在单一稳定结构,而是以大量不断变化的构象集合形式存在。传统结构预测方法难以描述这种高度异质的结构分布。研究人员结合物理力场模拟与多模态生成式深度学习,提出了名为 STARLING 的框架,可直接从蛋白序列快速生成高精度无序蛋白结构集合及其集合感知表示。该方法支持不同离子强度条件建模,并能通过贝叶斯最大熵重加权整合实验约束。STARLING不仅可用于结构预测,还能执行基于集合的相似蛋白搜索以及集合优先的序列设计,将候选设计时间从数周缩短到秒级。整体而言,该框架显著降低了研究无序蛋白生物物理行为的计算门槛,并展示了与实验数据良好一致性。

内在无序蛋白约占真核蛋白组的三成,它们在转录、信号传导等核心生物过程中发挥重要作用。由于缺乏固定三维结构,这类蛋白必须用“构象集合”而非单一结构来描述。虽然实验方法能够提供集合的部分信息,但难以完整获得所有构象分布,因此通常需要结合计算模拟。传统物理模拟方法虽然有效,但计算成本高、技术门槛大,即使粗粒化模拟也需较长时间。近年来深度学习显著推进了折叠蛋白结构预测,但这些方法通常优化单一结构输出,且依赖多序列比对信息,因此并不适用于无序蛋白。研究人员因此提出需要一种既快速又准确、能够直接从序列预测完整构象集合的新方法。
方法
研究人员将无序蛋白集合生成问题类比为文本生成多张图像的生成式AI任务,即同一输入应产生多个彼此独立但符合条件的结果。为实现这一目标,研究人员结合变分自编码器与扩散概率模型构建潜空间扩散模型。首先通过大规模粗粒化分子动力学模拟生成约数千万构象距离矩阵作为训练数据,这些距离矩阵被视为“图像”。随后使用VAE将高分辨率距离图压缩到低维潜空间,再在潜空间中训练扩散模型学习从噪声生成距离图的过程,并以蛋白序列和离子强度作为条件输入。最终模型由扩散网络与VAE解码器组成,可在推理阶段快速生成距离图并重建三维结构。该模型在GPU上可在十几秒内生成数百个构象,在普通CPU上亦可在数分钟完成。
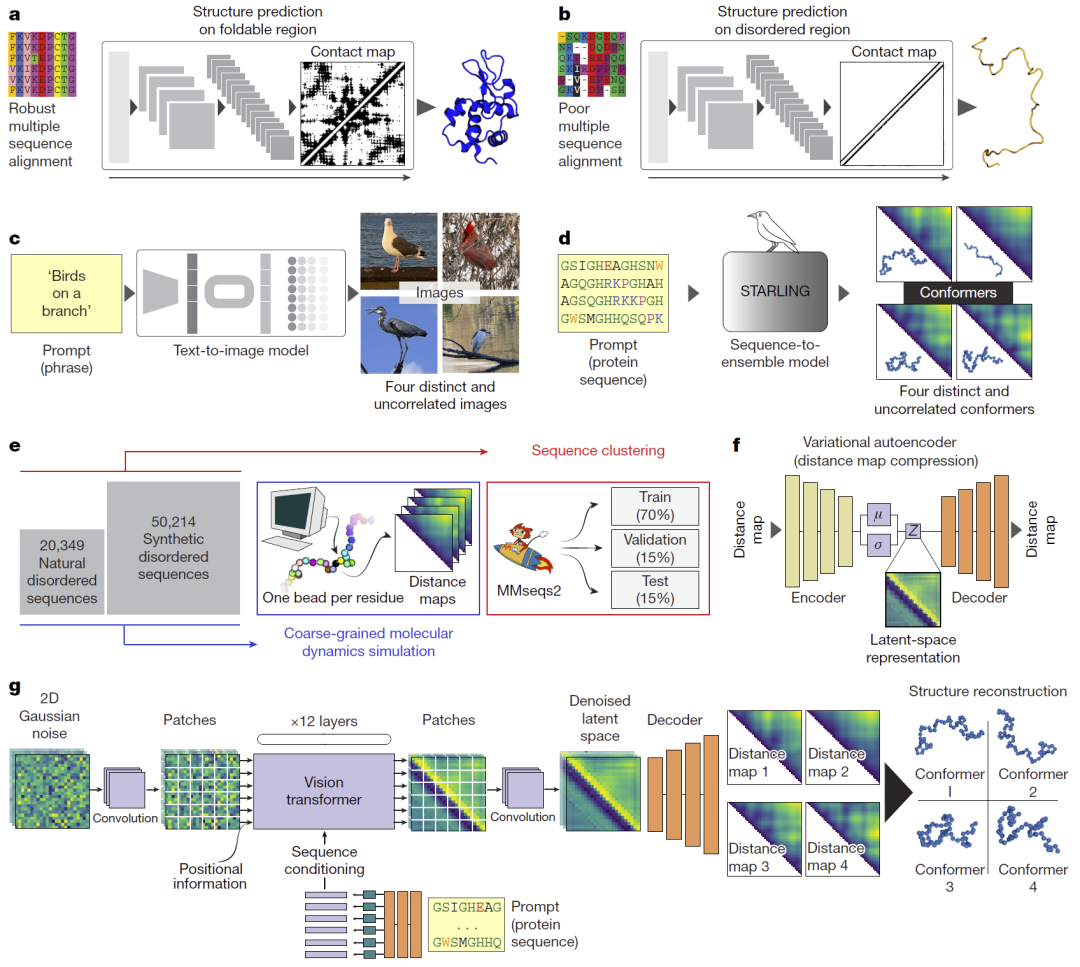
图1:STARLING方法框架与模型架构。
结果
模拟数据上的预测精度
研究人员首先在未见过的测试序列上评估模型预测的整体尺寸指标,例如回转半径和端到端距离。结果显示STARLING预测值与原始分子动力学模拟高度一致,误差极低。模型在不同离子强度条件下同样保持高精度,并且在长度匹配测试中证明模型确实学习到了序列化学组成与构象之间的真实关系,而非简单依赖链长。进一步分析显示STARLING不仅能预测平均值,还能准确再现完整分布及所有残基对之间的距离分布,证明其生成的集合具有真实结构统计特征。
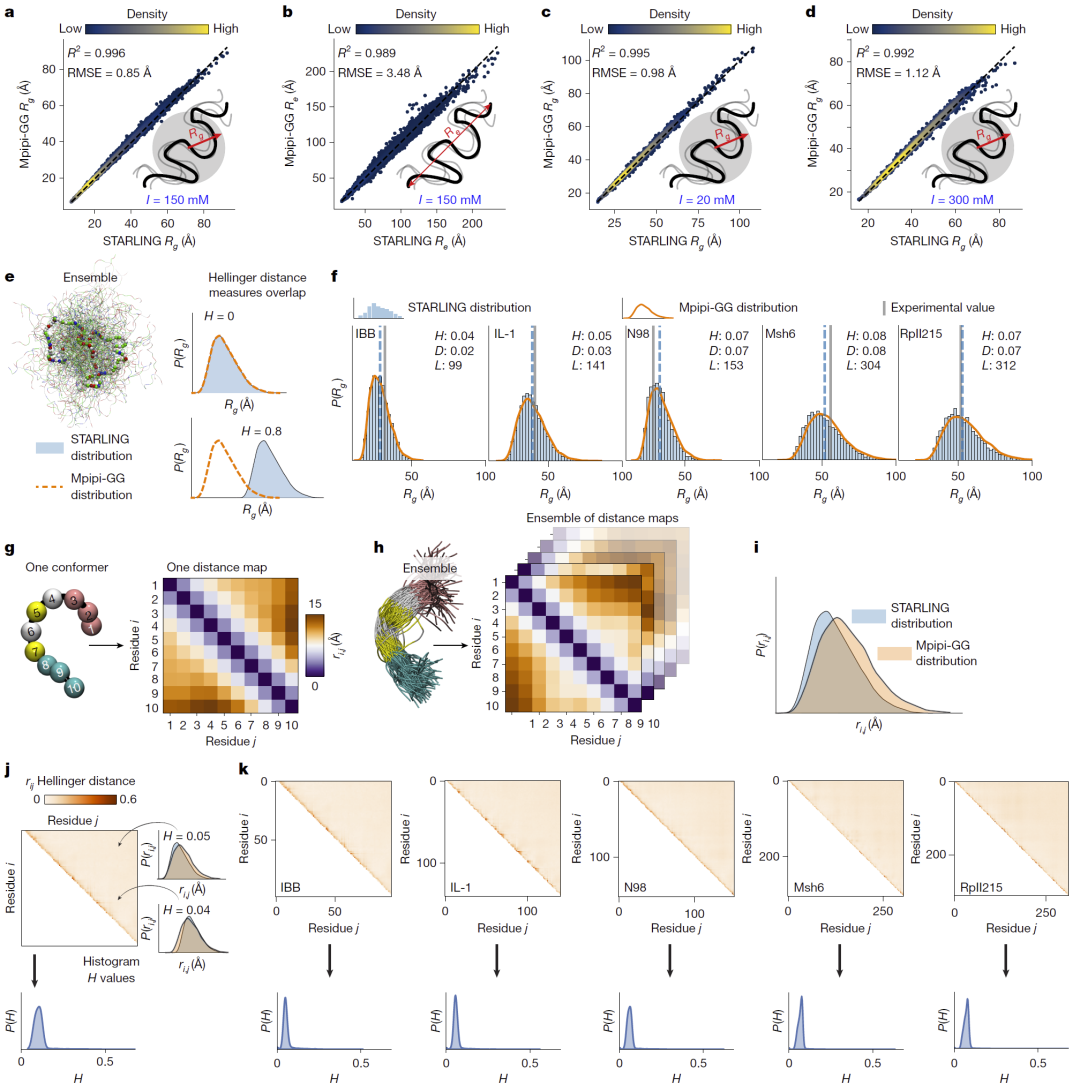
图2:在独立模拟测试数据上评估STARLING性能。
与实验数据一致性验证
研究人员利用包含一百余条序列的SAXS实验数据验证模型,发现预测的平均尺寸与实验结果高度一致。进一步通过从模型集合反向计算散射曲线并与真实实验曲线比较,结果在多种序列长度和化学组成范围内均表现出良好匹配。此外在单分子FRET实验测量的端到端距离数据上也获得相似精度,误差水平与不同实验团队之间的差异相当。这些结果表明STARLING能够可靠再现实验观测到的无序蛋白集合行为。
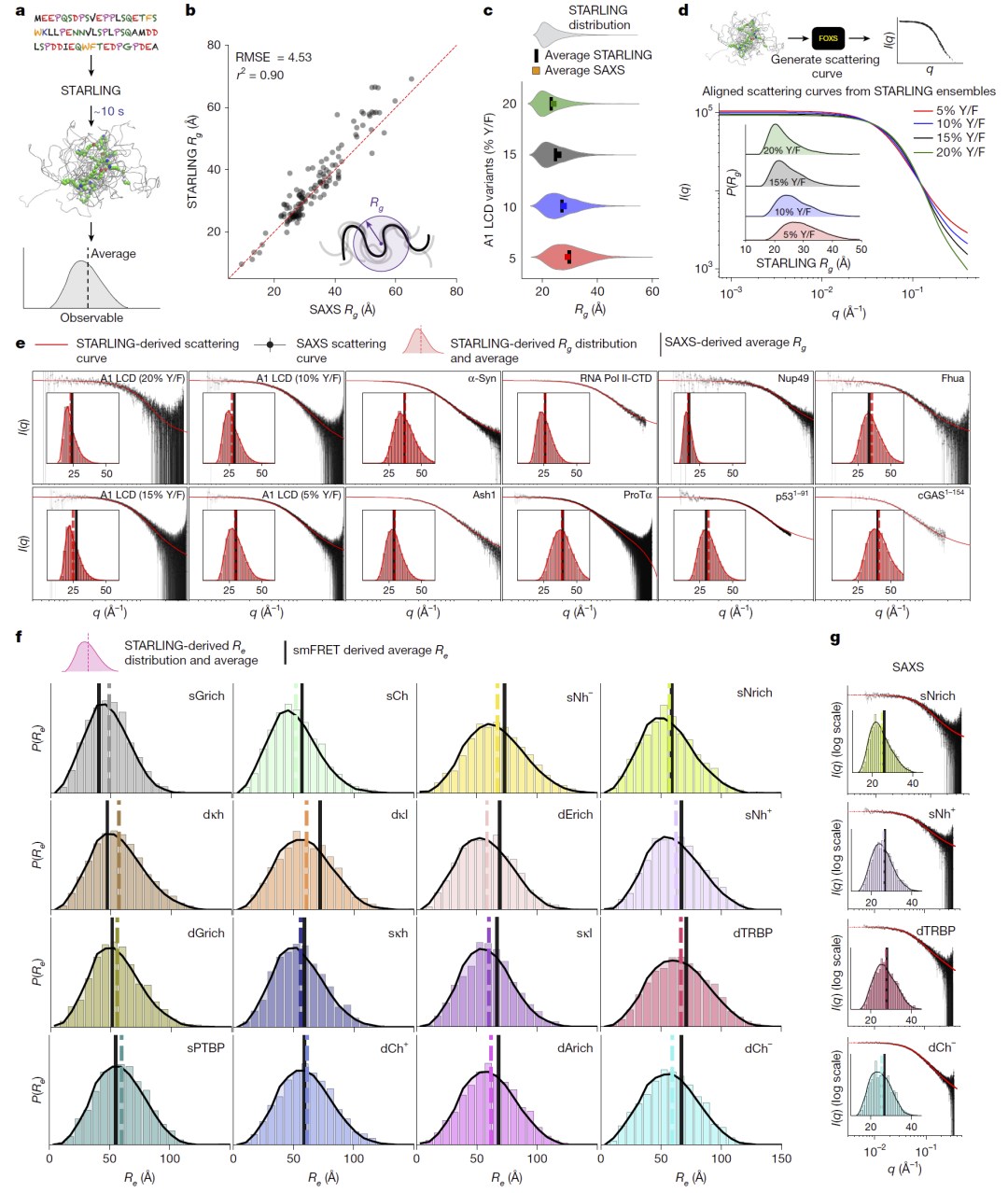
图3:STARLING的实验数据验证。
生物物理应用示例
研究人员随后展示了该模型在多个真实生物系统中的应用。例如在转录因子Myc的大型无序区中,模型预测出两个具有不同紧密程度的亚区域,并与已知功能结合区相对应。在RNA聚合酶II的CTD重复区中,模型与全原子模拟及实验结果一致,解释了其既具有扩展链统计行为又能参与相互作用的表面矛盾。研究人员还通过人为连接两个无序蛋白并预测其复合物集合,成功再现实验观测到的结合收缩行为,并在多个蛋白复合体系中获得一致结果。此外模型还能识别突变导致的长程相互作用变化,预测微蛋白集合特征,并实现根据目标集合性质进行序列反向设计。
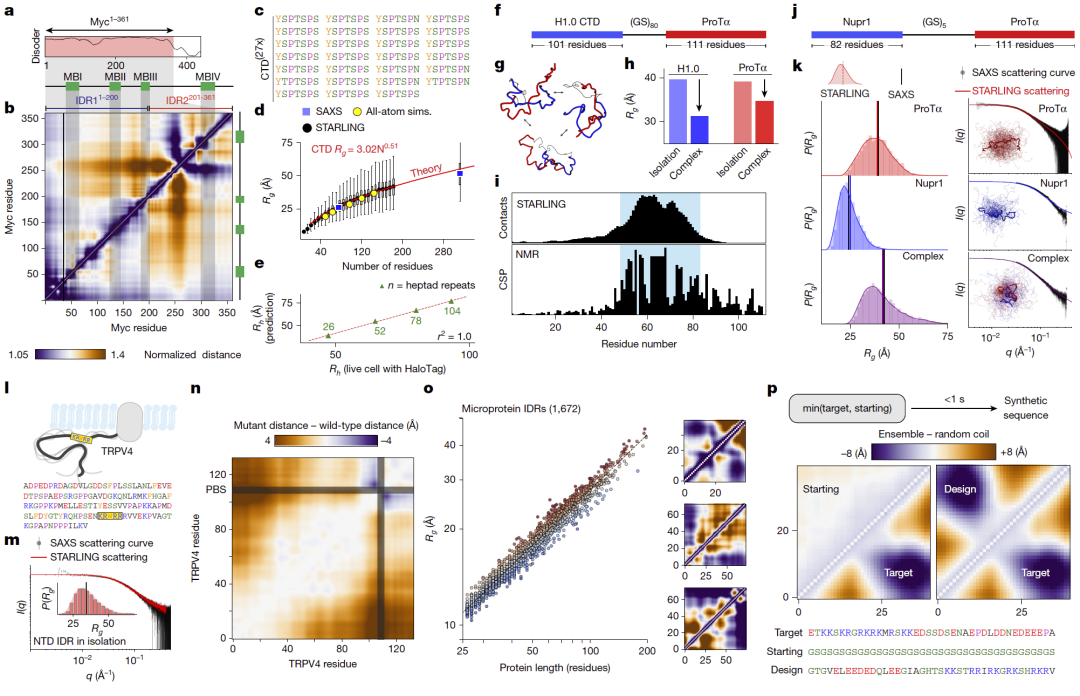
图4:利用STARLING进行生物物理表征与序列设计应用。
环境条件与搜索能力
由于模型在训练时考虑离子强度条件,因此能够预测不同盐浓度下的构象变化,并在未见过的中间条件上实现平滑插值。研究人员还引入贝叶斯最大熵重加权方法,将实验观测直接整合进集合中以提高一致性。此外STARLING还能生成集合感知的序列嵌入向量,使研究人员能够在数千万蛋白数据库中进行基于构象相似性的快速搜索,从而找到生物物理行为类似但序列差异较大的蛋白。
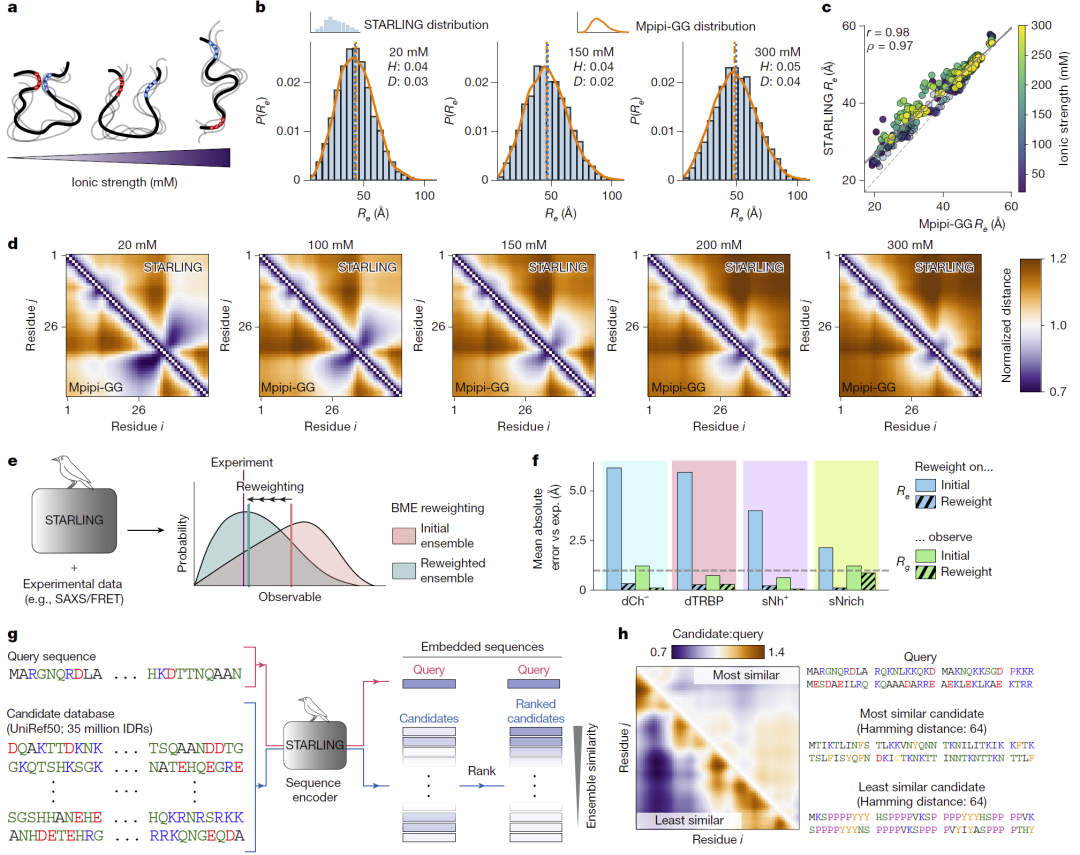
图5:STARLING实现溶液条件依赖的构象集合生成、贝叶斯重加权以及集合感知嵌入搜索。
讨论
研究人员认为STARLING的最大价值在于显著降低了研究无序蛋白构象集合的技术门槛,使任何研究人员都可以快速获得合理结构集合,并据此提出功能假设。与仅预测平均尺寸的模型相比,STARLING生成完整集合,使研究人员能够定位具体残基驱动的结构行为并与实验数据直接比较。该方法仍存在局限,例如基于粗粒化模型无法预测二级结构,也暂未考虑复杂环境因素或翻译后修饰,同时生成结果不包含时间动力学信息。然而其快速生成去相关构象的能力使其在长序列场景下具有明显优势,并可与分子动力学结合使用。总体而言,该框架为系统性研究“序列—集合—功能”关系以及大规模设计无序蛋白提供了新的可行路径。
整理 | DrugOne团队
参考资料
Novak, B., Lotthammer, J.M., Emenecker, R.J. et al. Accurate predictions of disordered protein ensembles with STARLING. Nature (2026).
https://doi.org/10.1038/s41586-026-10141-2

本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-02-25,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号