AI+Drug 文献速递 | 解锁蛋白-配体研究新方向:FDA 框架助力药物研发新征程

AI+Drug 文献速递 | 解锁蛋白-配体研究新方向:FDA 框架助力药物研发新征程

MindDance
发布于 2026-01-08 12:45:03
发布于 2026-01-08 12:45:03
1. A Folding-Docking-Affinity framework for protein-ligand binding affinity prediction
期刊: Communications Chemistry 链接: https://doi.org/10.1038/s42004-025-01506-1 代码: https://github.com/ZhiGroup/FDA
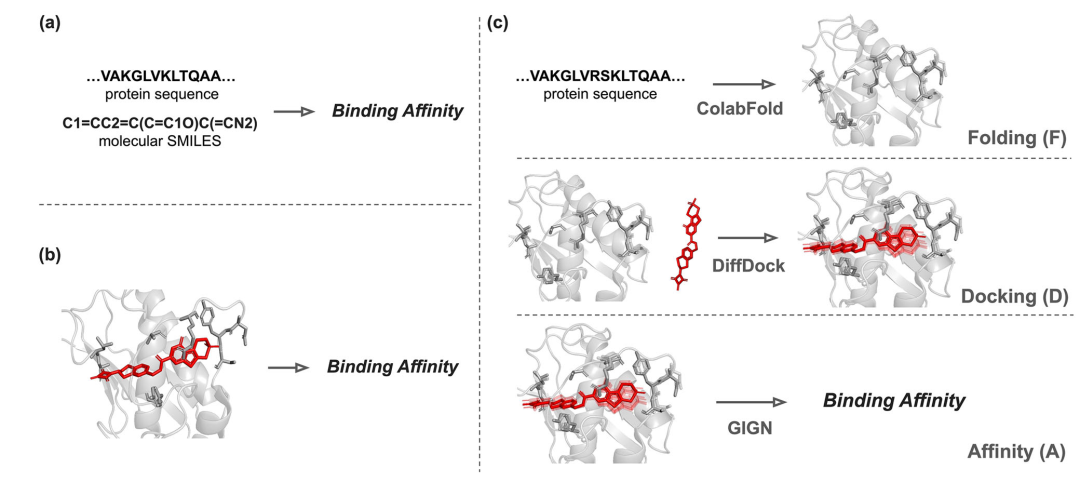
简介: 该论文提出Folding-Docking-Affinity (FDA)框架,通过计算蛋白质-配体结合构象预测结合亲和力,其组件可替换,适应相关方法发展。研究利用ColabFold、DiffDock和GIGN构建FDA框架,在DAVIS和KIBA两个激酶特异性数据集上进行基准测试,将数据集分为四种测试场景评估其性能,并开展消融实验和结合姿势增强实验。结果显示,FDA在多数场景下与最先进的无对接方法性能相当,在具有挑战性的“both-new”分裂中,DAVIS数据集的提高约32% ,KIBA数据集提高约12%,结合姿势增强能提升预测性能。该研究为结合结构用于准确预测结合亲和力提供了新方向。
2. FAIRSCAPE: An Evolving AI-readiness Framework for Biomedical Research
期刊: bioRxiv 链接: https://doi.org/10.1101/2024.12.23.629818
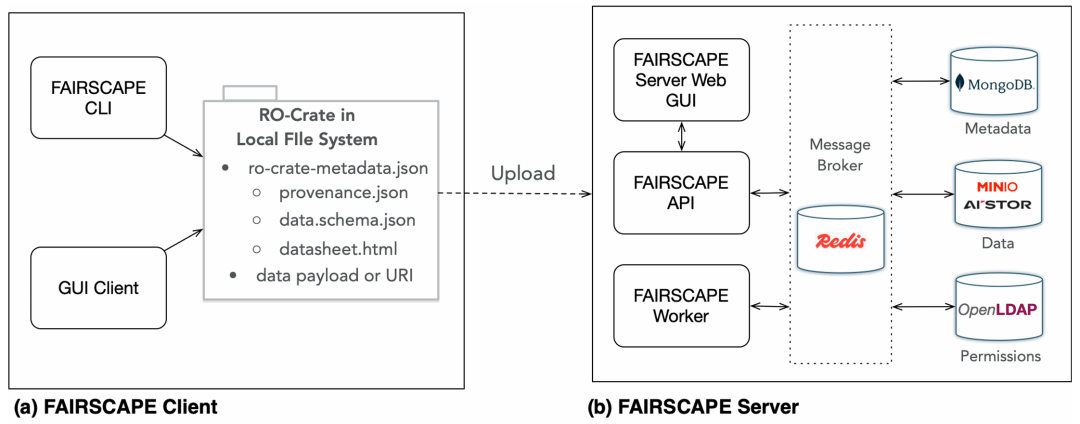
简介: 本文介绍了FAIRSCAPE框架,它能生成、整合关键的预模型XAI描述性元数据,助力生物医学数据集的AI应用,且符合FAIR原则。该框架由Python编写,包含客户端和服务器,可创建、管理、上传研究对象RO-Crate包。研究在服务器安装、配置,以及客户端安装、测试等方面进行实践,并展示了服务器Web GUI的功能。结果表明,FAIRSCAPE目前支持NIH Bridge2AI计划推荐的28个AI就绪标准中的27个。该框架为创建、管理和分发符合伦理、可用于AI的生物医学数据集提供了通用平台。
3. Machine learning: Python tools for studying biomolecules and drug design
期刊: Molecular Diversity 链接: https://doi.org/10.1007/s11030-025-11199-2
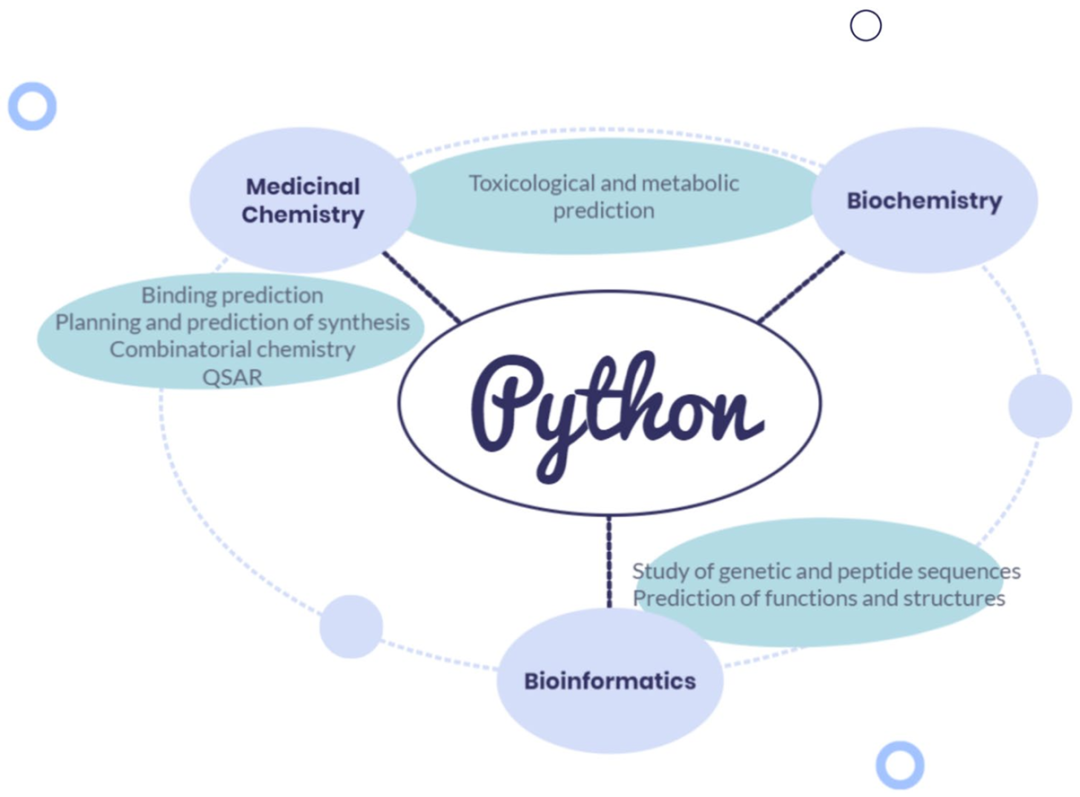
简介: 论文介绍了一系列用于生物分子研究和药物设计的Python工具,这些工具涵盖药物发现、高通量和虚拟筛选等多领域,拓展了研究方法。文中列举众多Python工具和库,如Biopython、RDKit等,从生物医学化学、生物信息学等多个方面阐述其功能及应用示例。实验涉及虚拟筛选、受体基于的药物设计等多种任务,使用了如DAVIS、KIBA等数据集。结果显示这些工具在各自应用场景中发挥了重要作用,提升了研究效率。该综述为科研人员在生物分子和药物设计研究中使用Python工具提供了参考,推动相关领域研究发展。
4. PoseX: AI Defeats Physics Approaches on Protein-Ligand Cross Docking
期刊: arXiv 链接: https://arxiv.org/abs/2505.01700v1 代码: https://github.com/CataAI/PoseX
简介: 论文提出PoseX这一开源基准,通过构建新数据集、纳入多种对接方法和设计新评估协议,对蛋白-配体对接算法进行全面评估,发现AI对接方法在整体上优于传统方法,且经优化后性能更优。研究整合22种对接方法,涵盖传统物理方法、AI对接方法和AI共折叠方法,使用自建的PoseX数据集(含718个自对接和1312个交叉对接条目)和Astex数据集进行实验,以TM分数、RMSD等作为评估指标。结果显示,AI对接方法在整体RMSD性能上领先,如Uni-Mol和SurfDock在Astex基准测试中成功率超90%;结合优化方法后AI模型表现最佳;口袋感知模型精度更高;大型共折叠模型有竞争力但存在局限。该研究为蛋白-配体对接算法评估提供了全面基准,推动了相关领域发展。

5. MHNfs: Prompting In-Context Bioactivity Predictions for Low-Data Drug Discovery
期刊: Journal of Chemical Information and Modeling 链接: https://doi.org/10.1021/acs.jcim.4c02373 代码: https://github.com/ml-jku/MHNfs
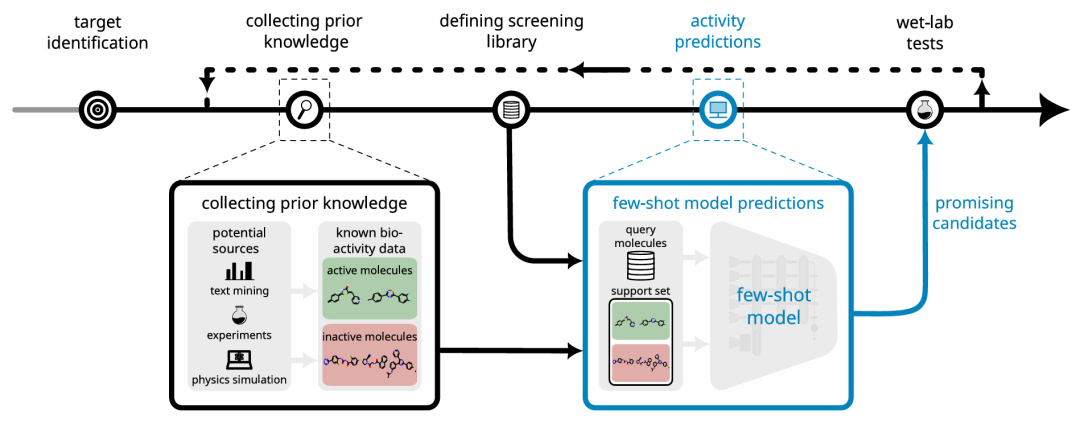
简介: 论文介绍了MHNfs应用程序,它利用先进的少样本学习模型,通过简单交互界面实现低数据场景下分子活性预测,为药物发现提供了有效工具。该应用基于MHNfs模型,通过上下文模块、交叉注意力模块和相似性模块,利用少量已知活性和非活性分子进行预测。研究人员使用FS-Mol数据集训练模型,并在PubChem数据集上进行评估,将PubChem生物测定数据重铸为少样本预测任务,与随机森林基线模型对比。结果表明,MHNfs在几乎所有测试场景和指标上显著优于随机森林基线,在支持集较小(如4个活性和4个非活性分子)时,ROC-AUC中位数已超0.7。该应用为药物发现早期阶段的分子活性预测提供了便捷且有效的手段。
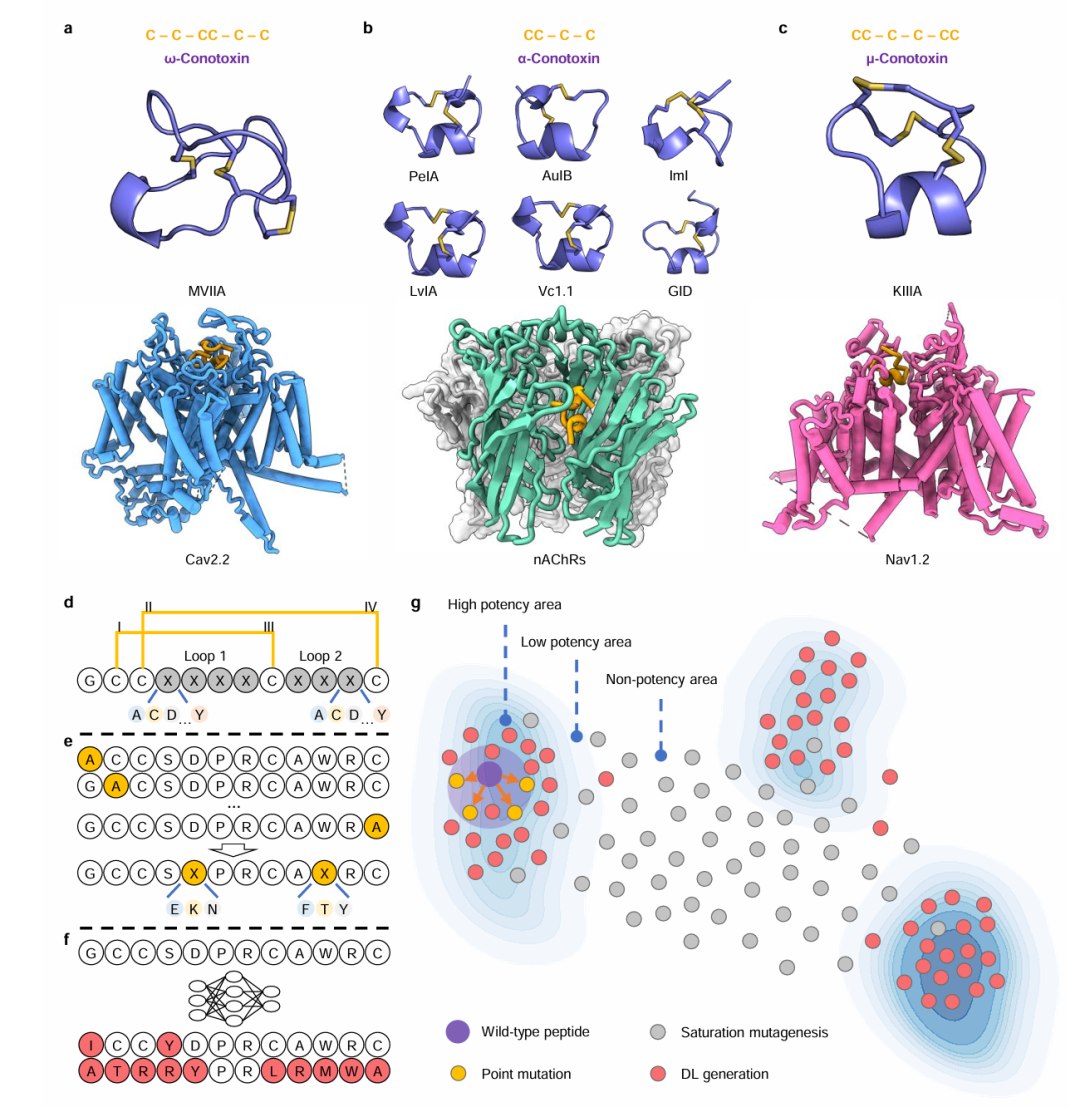
6. CreoPep: A Universal Deep Learning Framework for Target-Specific Peptide Design and Optimization
期刊: arXiv 链接: https://arxiv.org/abs/2505.02887v1 代码: https://github.com/gc-js/CreoPep https://huggingface.co/spaces/oucgc1996/CreoPep
简介: 论文提出CreoPep,这一深度学习框架整合了掩码语言建模和渐进式掩码策略,用于设计高亲和力肽突变体,加速了靶向特异性肽的发现。该框架基于ProtBert的掩码语言建模框架,通过渐进式掩码策略、多任务学习和数据增强等创新设计,生成结构和功能多样的肽。研究人员收集2088种芋螺毒素构建数据集,经多轮FoldX筛选和温度控制的多项式采样进行数据增强,训练CreoPep模型。以ImI/α7 nAChR系统为例,实验验证了模型生成的突变体。结果显示,13个候选突变体中有7个对hα7 nAChR有显著抑制活性,如CP_α7_1和CP_α7_6的 (IC50) 值分别为405.1 nM和504.6 nM。该研究为靶向特异性肽的设计和优化提供了通用且强大的平台。
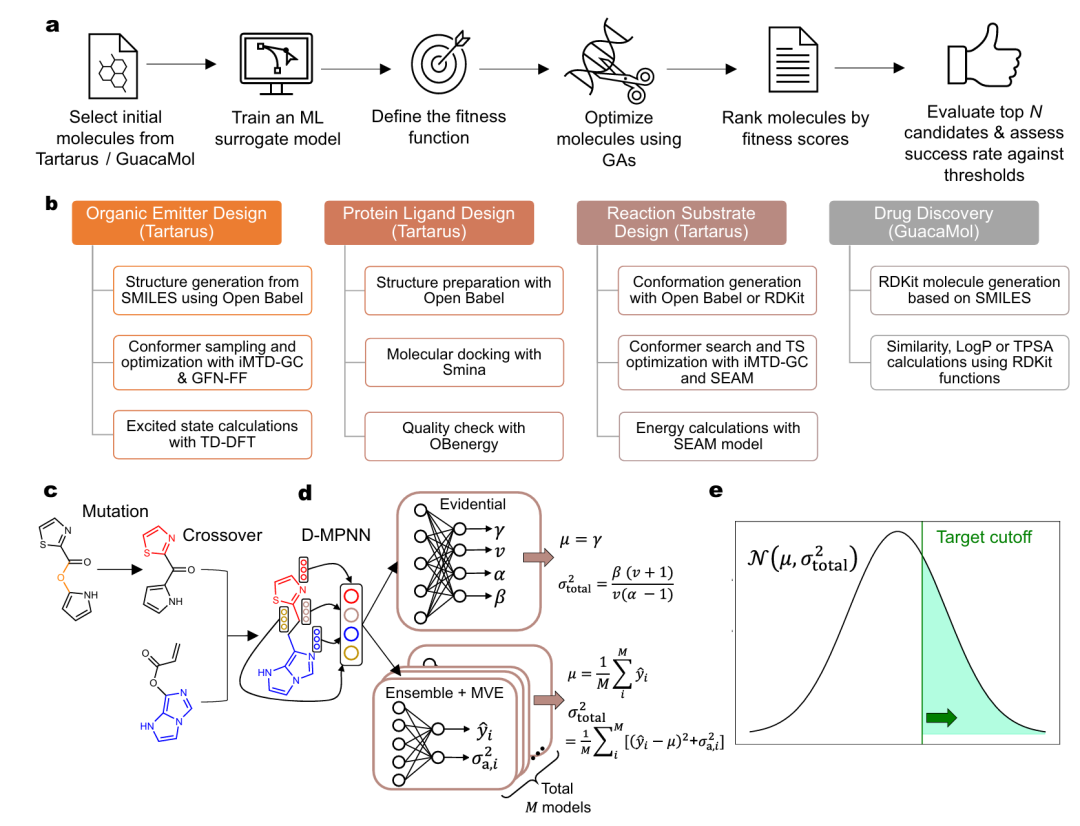
7. Uncertainty quantification with graph neural networks for efficient molecular design
期刊: Nature Communications 链接: https://doi.org/10.1038/s41467-025-58503-0 代码: https://doi.org/10.5281/zenodo.14729022
简介: 该论文将不确定性量化(UQ)、定向消息传递神经网络(D-MPNNs)和遗传算法(GAs)相结合,解决分子设计在化学空间优化中维持预测准确性的挑战,发现概率改进优化(PIO)方法在多数情况下能提升优化成功率。研究构建了基于D-MPNN的代理模型,利用Tartarus和GuacaMol平台的数据集,通过对比直接目标最大化(DOM)、预期改进(EI)和PIO等方法进行实验。结果显示,在单目标任务中,PIO方法在多数任务上命中率最高;在多目标任务中,PIO也表现出色,能更好地平衡多个目标,优于其他方法。该研究为计算辅助分子设计中整合UQ提供了实用指导,推动了分子设计领域的发展。
8. Enhancing Chemical Reaction and Retrosynthesis Prediction with Large Language Model and Dual-task Learning
期刊: arXiv 链接: https://arxiv.org/abs/2505.02639v1
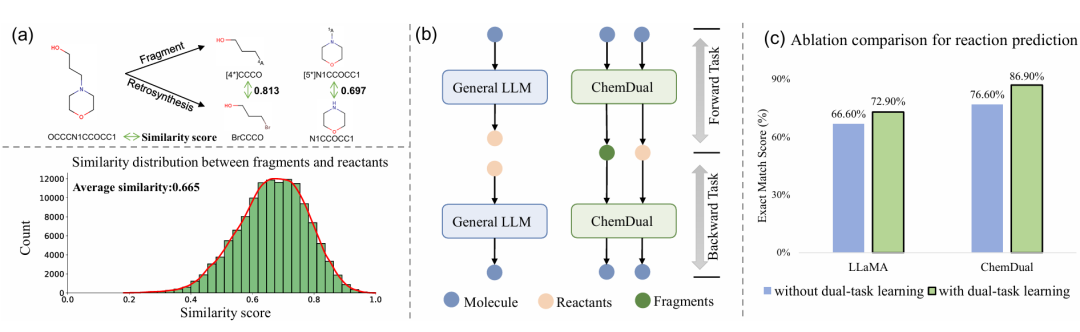
简介: 论文提出ChemDual框架,通过构建大规模数据集和采用双任务学习策略,提升大语言模型在化学反应和逆合成预测任务中的性能。该框架基于LLaMA,利用多尺度分词器和双任务学习,将分子的反应和逆合成视为相关过程进行联合优化。研究人员从ChEMBL-34数据库收集数据构建440万分子的数据集,在Mol-Instruction、USPTO-50K和ChemLLMBench等数据集上,将ChemDual与多种基线模型对比。结果表明,ChemDual在反应和逆合成预测任务上均取得最先进的成绩,在多个评估指标上优于其他模型,且生成的化合物与目标蛋白结合亲和力强。该研究为化学反应和逆合成预测提供了更有效的方法,展现了在药物设计中的潜力。
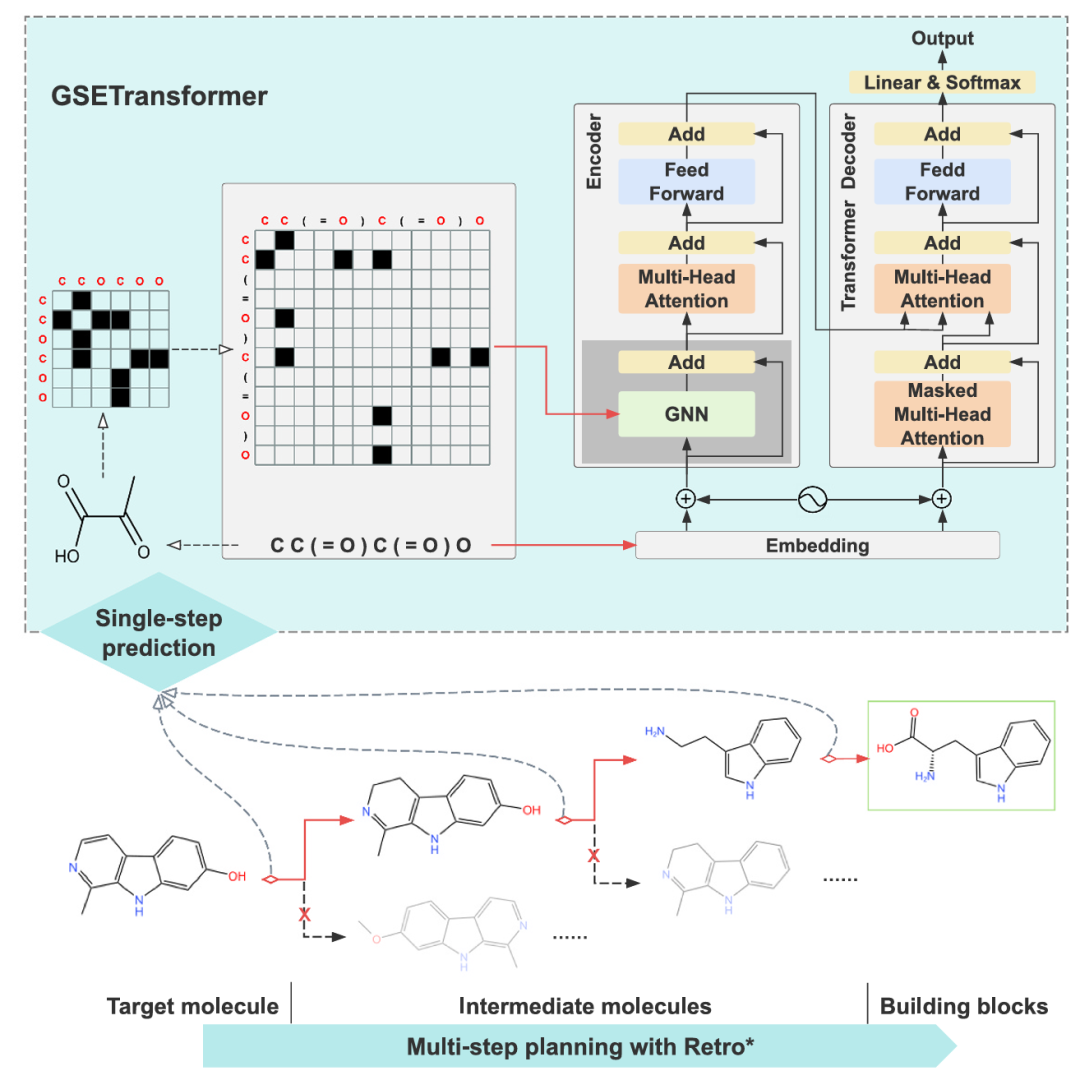
9. Graph-sequence enhanced transformer for template-free prediction of natural product biosynthesis
期刊: Patterns 链接: https://doi.org/10.1016/j.patter.2025.101259 代码: https://github.com/momozhangcn/GSETRetro https://doi.org/10.5281/zenodo.15023160
简介: 论文提出GSETransformer框架,将图神经网络与序列Transformer结合,用于天然产物生物合成的无模板预测,在基准评估中表现优异,并提供了用户友好的图形界面。该框架基于标准的编码器-解码器架构,在编码器中融入图神经网络处理分子图结构信息。研究使用USPTO-50K、BioChem Plus等数据集进行实验,对比多种无模板逆合成方法。结果显示,在单步逆合成任务上,GSETransformer在BioChem Plus数据集的top-3、top-5和top-10准确率表现最佳;在多步逆合成任务中,其成功率和路径命中率等指标也表现出色。此外,开发的GUI软件集成了酶预测和ADMET属性预测功能。该研究为天然产物生物合成预测提供了有效工具,推动了计算生物合成领域的发展。
10. BOOM: Benchmarking Out-Of-distribution Molecular Property Predictions of Machine Learning Models
期刊: arXiv 链接: https://arxiv.org/abs/2505.01912v1
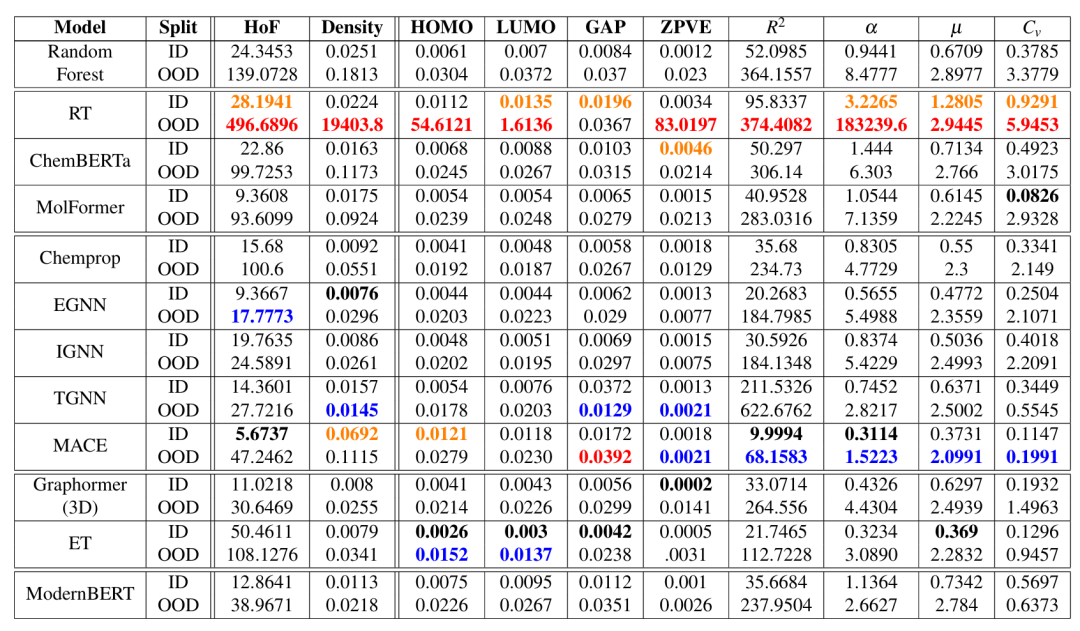
简介: 文章提出BOOM基准测试,用于评估机器学习模型在分子属性预测中的分布外(OOD)性能,发现现有模型在OOD任务上表现不佳,并探讨了影响OOD性能的因素。研究收集10个分子属性数据集,包括QM9和10k数据集,将模型输出中属性值的尾部分布作为OOD测试集,评估了12种模型。实验结果表明,没有模型能在所有OOD任务中表现出色,即使表现最佳的模型,其平均OOD误差也比分布内高出3倍;预训练对提升OOD性能效果不明显,特定任务相关的预训练才有帮助;超参数优化对部分简单属性有一定效果;3D模型在多数任务上优于基于SMILES的模型;数据增强可提高部分任务的泛化能力。该研究为化学领域的OOD研究提供了基准,指明了提升模型OOD性能的方向。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-05-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号