Signac R|如何合并多个 Seurat 对象 (2)

Signac R|如何合并多个 Seurat 对象 (2)

数据科学工厂
发布于 2024-12-30 09:54:43
发布于 2024-12-30 09:54:43
引言
在本文中演示了如何合并包含单细胞染色质数据的多个 Seurat 对象。为了进行演示,将使用 10x Genomics 提供的四个 scATAC-seq PBMC 数据集:
- 500-cell PBMC
- 1k-cell PBMC
- 5k-cell PBMC
- 10k-cell PBMC
构建数据对象
接下来,将利用已经量化的矩阵数据,针对每个数据集构建一个 Seurat 数据对象。在这个过程中,每个数据集对应的片段对象(Fragment 对象)将被妥善保存在相应的分析模块(assay)里。
pbmc500_assay <- CreateChromatinAssay(pbmc500.counts, fragments = frags.500)
pbmc500 <- CreateSeuratObject(pbmc500_assay, assay = "ATAC", meta.data=md.500)
pbmc1k_assay <- CreateChromatinAssay(pbmc1k.counts, fragments = frags.1k)
pbmc1k <- CreateSeuratObject(pbmc1k_assay, assay = "ATAC", meta.data=md.1k)
pbmc5k_assay <- CreateChromatinAssay(pbmc5k.counts, fragments = frags.5k)
pbmc5k <- CreateSeuratObject(pbmc5k_assay, assay = "ATAC", meta.data=md.5k)
pbmc10k_assay <- CreateChromatinAssay(pbmc10k.counts, fragments = frags.10k)
pbmc10k <- CreateSeuratObject(pbmc10k_assay, assay = "ATAC", meta.data=md.10k)
整合数据对象
既然每个数据对象都包含了一套相同的特征分析(assay),就可以使用常规的合并功能来整合它们。这个过程还会整合所有的片段对象,确保在最终整合后的数据对象中,每个细胞的片段信息得以完整保留。
# add information to identify dataset of origin
pbmc500$dataset <- 'pbmc500'
pbmc1k$dataset <- 'pbmc1k'
pbmc5k$dataset <- 'pbmc5k'
pbmc10k$dataset <- 'pbmc10k'
# merge all datasets, adding a cell ID to make sure cell names are unique
combined <- merge(
x = pbmc500,
y = list(pbmc1k, pbmc5k, pbmc10k),
add.cell.ids = c("500", "1k", "5k", "10k")
)
combined[["ATAC"]]
## ChromatinAssay data with 89951 features for 21688 cells
## Variable features: 0
## Genome:
## Annotation present: FALSE
## Motifs present: FALSE
## Fragment files: 4
combined <- RunTFIDF(combined)
combined <- FindTopFeatures(combined, min.cutoff = 20)
combined <- RunSVD(combined)
combined <- RunUMAP(combined, dims = 2:50, reduction = 'lsi')
DimPlot(combined, group.by = 'dataset', pt.size = 0.1)
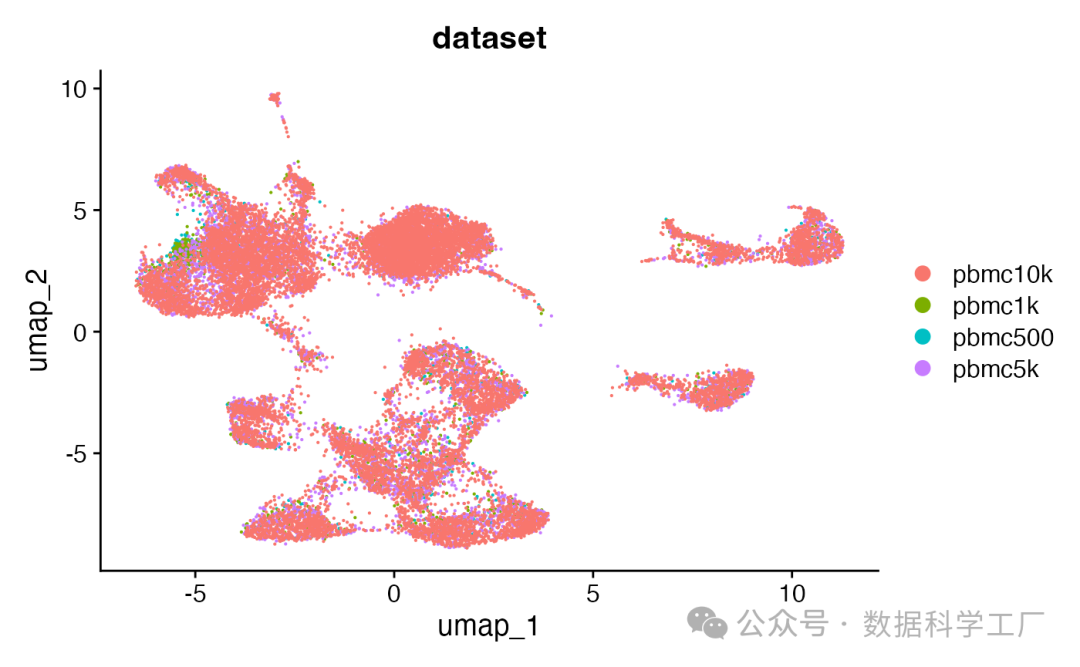
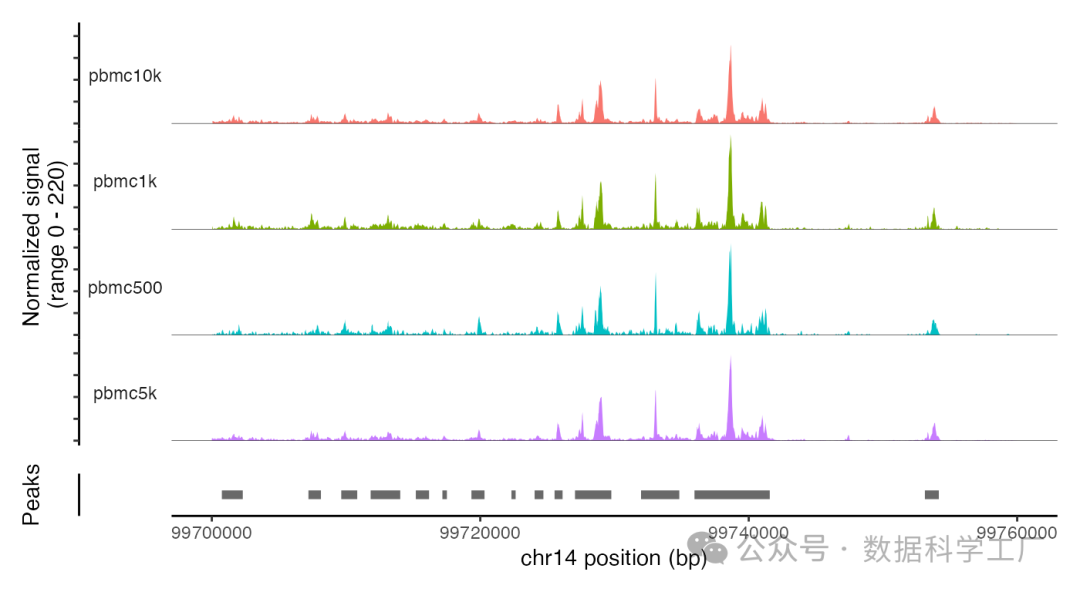
整合后的数据对象整合了全部四个片段对象,并且在其内部建立了一个细胞名称的映射机制,将对象内的细胞名称与各个片段文件中的细胞名称相对应。这样一来,就能够直接从这些文件中提取信息,而无需对每个片段文件中的细胞名称进行修改。为了验证从片段文件中提取数据的功能是否在整合后的对象上正常运作,可以通过绘制基因组中的特定区域来进行检验。
CoveragePlot(
object = combined,
group.by = 'dataset',
region = "chr14-99700000-99760000"
)
合并数据
之前讨论的方法需要能够获取到每个数据集对应的片段文件。然而,在某些情况下,这些数据可能不可用(尽管可以通过 sinto 工具从 BAM 文件生成片段文件)。即便如此,还是能够构建一个合并后的数据对象,但要意识到,最终得到的合并计数矩阵可能达不到理想的精确度。
在 Signac 中,针对 ChromatinAssay 对象的合并函数会将相互重叠的峰视为相同的,并调整这些峰所跨越的基因组区域,以确保合并过程中的每个对象中的特征保持一致。需要指出的是,这种做法可能会导致计数矩阵出现一些误差,因为部分峰的范围可能会被扩展,覆盖到原本未被量化的区域。在无法重新量化的情况下,这是能够采取的最佳方案,建议在可能的情况下,始终按照上述的合并步骤进行操作。
接下来,将演示如何在不建立统一特征集的前提下,合并四个相同的 PBMC 数据集。
# load the count matrix for each object that was generated by cellranger
counts.500 <- Read10X_h5("pbmc500/atac_pbmc_500_nextgem_filtered_peak_bc_matrix.h5")
counts.1k <- Read10X_h5("pbmc1k/atac_pbmc_1k_nextgem_filtered_peak_bc_matrix.h5")
counts.5k <- Read10X_h5("pbmc5k/atac_pbmc_5k_nextgem_filtered_peak_bc_matrix.h5")
counts.10k <- Read10X_h5("pbmc10k/atac_pbmc_10k_nextgem_filtered_peak_bc_matrix.h5")
# create objects
pbmc500_assay <- CreateChromatinAssay(counts = counts.500, sep = c(":", "-"), min.features = 500)
pbmc500 <- CreateSeuratObject(pbmc500_assay, assay = "peaks")
pbmc1k_assay <- CreateChromatinAssay(counts = counts.1k, sep = c(":", "-"), min.features = 500)
pbmc1k <- CreateSeuratObject(pbmc1k_assay, assay = "peaks")
pbmc5k_assay <- CreateChromatinAssay(counts = counts.5k, sep = c(":", "-"), min.features = 500)
pbmc5k <- CreateSeuratObject(pbmc5k_assay, assay = "peaks")
pbmc10k_assay <- CreateChromatinAssay(counts = counts.10k, sep = c(":", "-"), min.features = 1000)
pbmc10k <- CreateSeuratObject(pbmc10k_assay, assay = "peaks")
# add information to identify dataset of origin
pbmc500$dataset <- 'pbmc500'
pbmc1k$dataset <- 'pbmc1k'
pbmc5k$dataset <- 'pbmc5k'
pbmc10k$dataset <- 'pbmc10k'
# merge
combined <- merge(
x = pbmc500,
y = list(pbmc1k, pbmc5k, pbmc10k),
add.cell.ids = c("500", "1k", "5k", "10k")
)
# process
combined <- RunTFIDF(combined)
combined <- FindTopFeatures(combined, min.cutoff = 20)
combined <- RunSVD(combined)
combined <- RunUMAP(combined, dims = 2:50, reduction = 'lsi')
DimPlot(combined, group.by = 'dataset', pt.size = 0.1)
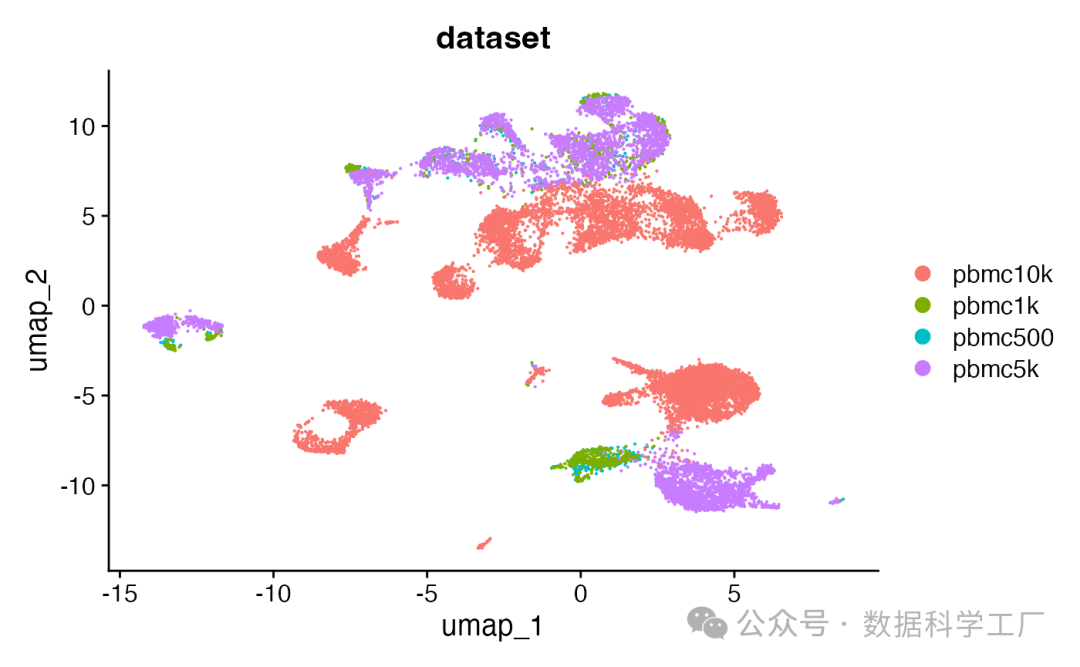
总结
本文[1]提供了一个详细的流程来合并单细胞染色质数据集,包括数据下载、预处理、合并以及后续的分析和可视化步骤。强调了在合并过程中创建共有峰值集合的重要性,并提供了在没有片段文件时的替代方法。
Reference
[1]
Source: https://stuartlab.org/signac/articles/merging
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-09-06,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号