scRNA|R版CytoTRACE v2从0开始完成单细胞分化潜能预测

scRNA|R版CytoTRACE v2从0开始完成单细胞分化潜能预测

生信补给站
发布于 2024-04-26 11:15:54
发布于 2024-04-26 11:15:54
CytoTRACE v2 在2024.03月发表在预印本Mapping single-cell developmental potential in health and disease with interpretable deep learning。V2 使用可解释性的AI算法来预测单细胞RNA测序数据的细胞分化潜能。除了给出从0(分化)到1(全能)的连续发育潜能度量结果外,还根据细胞的发育潜能进行分为6类:具有广泛分化潜能的全能(totipotent)和多能(pluripotent)干细胞,到能够产生不同数量的下游细胞类型的 谱系限制性多能细胞(lineage-restricted oligopotent),多能(multipotent)和单能(unipotent)细胞,再到最终的 分化(differentiated)细胞。
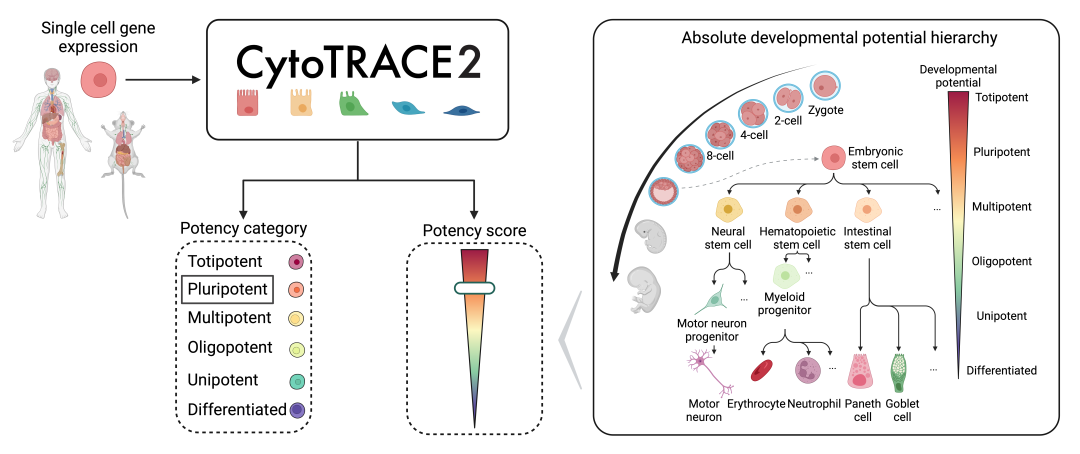
相较V1的功能和理论的改进详见文献正文,在代码实现上CytoTRACE v2中拆分为了R版本和Python版本,安装R版本的话无需配置python的环境,使用门槛大幅降低。
一 载入R包,数据
1,R包安装 及 解决报错
根据https://github.com/digitalcytometry/cytotrace2?tab=readme-ov-file中的方式进行安装
(1)使用devtools::install_github直接安装
devtools::install_github("digitalcytometry/cytotrace2", subdir = "cytotrace2_r")
library(CytoTRACE2)
# 出现报错
Using github PAT from envvar GITHUB_TOKEN
Downloading GitHub repo digitalcytometry/cytotrace2@HEAD
Error in utils::download.file(url, path, method = method, quiet = quiet, :
download from 'https://api.github.com/repos/digitalcytometry/cytotrace2/tarball/HEAD' failed(2)如果出现上述的报错,这时候只要将报错内容的“https://api.github.com/repos/digitalcytometry/cytotrace2/tarball/HEAD” 复制到网址搜索栏回车,就会下载一个文件tar.gz的压缩文件,然后我们再本地安装即可。
# 本地安装
remotes::install_local("./digitalcytometry-cytotrace2-6fe2bad.tar.gz",
subdir = "cytotrace2_r", # 特殊的
upgrade = F,dependencies = T)
library(CytoTRACE2)
library(tidyverse)
library(Seurat)注:打开tar.gz压缩包可以看到作者分的python 和r 版本,所以这里需要使用subdir参数指定为cytotrace2_r 。
注:其他的github包出现类型报错也可以使用上述方式进行解决,一般不需要设置subdir 。
2,准备单细胞数据
然后使用之前注释过的sce.anno.RData数据 ,为节省资源,每种细胞类型随机抽取30%的数据。
load("sce.anno.RData")
sce2@meta.data$CB <- rownames(sce2@meta.data)
sample_CB <- sce2@meta.data %>%
group_by(celltype) %>%
sample_frac(0.3)
sce3 <- subset(sce2,CB %in% sample_CB$CB)
sce3
# An object of class Seurat
二 CytoTRACE v2 分析
1,CytoTRACE v2 分析
该版本可以接受单细胞对象 或者 单细胞矩阵的两种形式,物种可以是人或者小鼠(默认)。本推文是使用 人 的单细胞对象(sce3)进行cytotrace2分析的示例。
#######输入seurat 对象###########
cytotrace2_result_sce <- cytotrace2(sce3,
is_seurat = TRUE,
slot_type = "counts",
species = 'human',
seed = 1234)
cytotrace2_result_sce
An object of class Seurat
51911 features across 4202 samples within 1 assay
Active assay: RNA (51911 features, 2000 variable features)
4 dimensional reductions calculated: pca, umap, tsne, harmony输入的是单细胞对象,得到的也是单细胞对象,且meta信息中包含了相关score的结果。
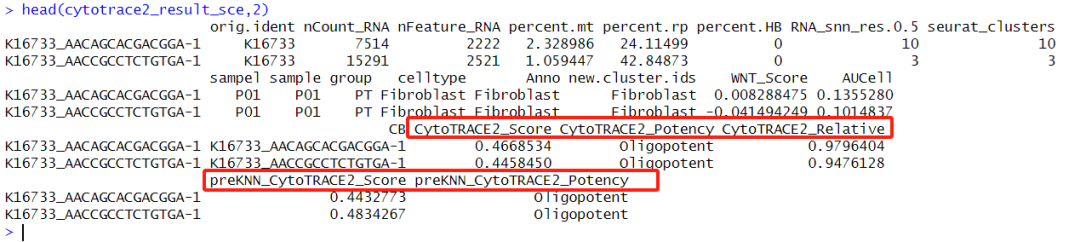
其中CytoTRACE2_Relative为score的具体数值结果;CytoTRACE2_Potency为文章开头提到的的六类结果。
注1:cytotrace2默认的是小鼠,所以需要指定species = 'human' ;如果是单细胞对象的话需要指定is_seurat = TRUE ;指定seed 方便后续的结果复现。。
2,CytoTRACE v2可视化
(1)v2在 plotData
同cytotrace v1的可视化函数不一样,v2在 plotData函数中包装了一些常见的可视化结果 ,可以先设定待展示的表型(celltype) 。
# making an annotation dataframe that matches input requirements for plotData function
annotation <- data.frame(phenotype = sce3@meta.data$celltype) %>%
set_rownames(., colnames(sce3))
# plotting
plots <- plotData(cytotrace2_result = cytotrace2_result_sce,
annotation = annotation,
is_seurat = TRUE)
# 绘制CytoTRACE2_Potency的umap图
p1 <- plots$CytoTRACE2_UMAP
# 绘制CytoTRACE2_Potency的umap图
p2 <- plots$CytoTRACE2_Potency_UMAP
# 绘制CytoTRACE2_Relative的umap图 ,v1
p3 <- plots$CytoTRACE2_Relative_UMAP
# 绘制各细胞类型CytoTRACE2_Score的箱线图
p4 <- plots$CytoTRACE2_Boxplot_byPheno
(p1+p2+p3+p4) + plot_layout(ncol = 2)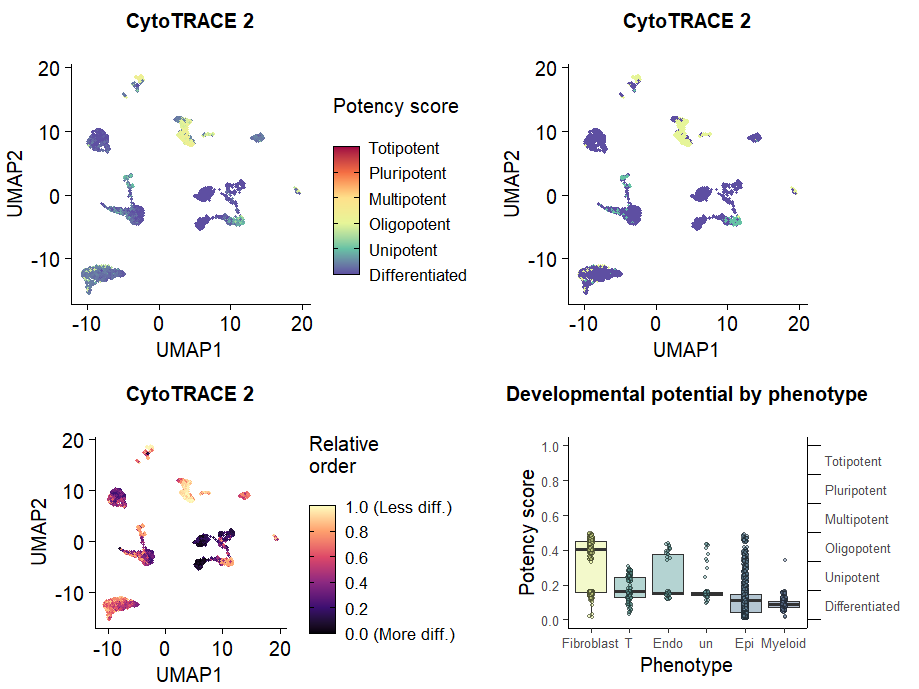
(2)调整出图的风格,与V1接近(plotData函数中的代码)
FeaturePlot(cytotrace2_result_sce, "CytoTRACE2_Relative",pt.size = 1.5) +
scale_colour_gradientn(colours =
(c("#9E0142", "#F46D43", "#FEE08B", "#E6F598",
"#66C2A5", "#5E4FA2")),
na.value = "transparent",
limits = c(0, 1),
breaks = seq(0, 1, by = 0.2),
labels = c("0.0 (More diff.)",
"0.2", "0.4", "0.6", "0.8", "1.0 (Less diff.)"),
name = "Relative\norder \n",
guide = guide_colorbar(frame.colour = "black",
ticks.colour = "black")) +
ggtitle("CytoTRACE 2") +
xlab("UMAP1") + ylab("UMAP2") +
theme(legend.text = element_text(size = 10),
legend.title = element_text(size = 12),
axis.text = element_text(size = 12),
axis.title = element_text(size = 12),
plot.title = element_text(size = 12,
face = "bold", hjust = 0.5,
margin = margin(b = 20))) +
theme(aspect.ratio = 1)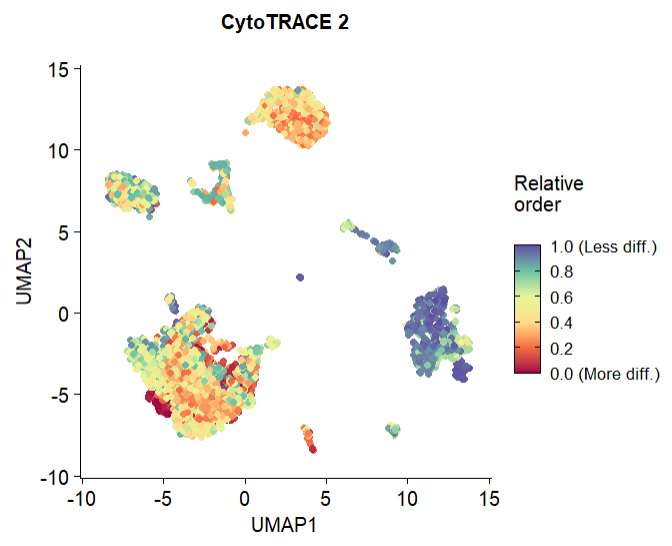
单细胞的很多可视化都是可以使用ggplot2进行自定义的。更多ggplot2 的调整可以参考ggplot2 | 关于标题,坐标轴和图例的细节修改,你可能想了解,ggplot2|详解八大基本绘图要素,ggplot2|theme主题设置,详解绘图优化-“精雕细琢” 等 。
(3)细胞类型-箱线图
除了p4自带的箱线图,也可以根据需求自行绘制 scRNA分析|使用AddModuleScore 和 AUcell进行基因集打分,可视化
library(ggpubr)
p1 <- ggboxplot(cytotrace2_result_sce@meta.data, x="celltype", y="CytoTRACE2_Score", width = 0.6,
color = "black",#轮廓颜色
fill="celltype",#填充
palette = "npg",
xlab = F, #不显示x轴的标签
bxp.errorbar=T,#显示误差条
bxp.errorbar.width=0.5, #误差条大小
size=1, #箱型图边线的粗细
outlier.shape=NA, #不显示outlier
legend = "right") #图例放右边
###指定组比较
my_comparisons <- list(c("Epi", "un"), c("T", "un"),c("Myeloid", "un"))
p1+stat_compare_means(comparisons = my_comparisons,
method = "wilcox.test")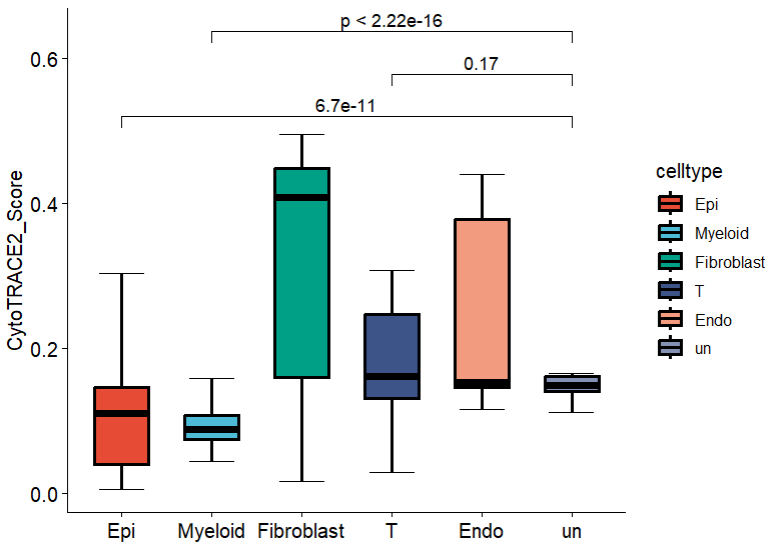
3,结合monocle2 确定起点
相关的预测结果已经在metadata中了,可以在monocle2中绘制基于分化 score的结果,以此来帮助确定起点。
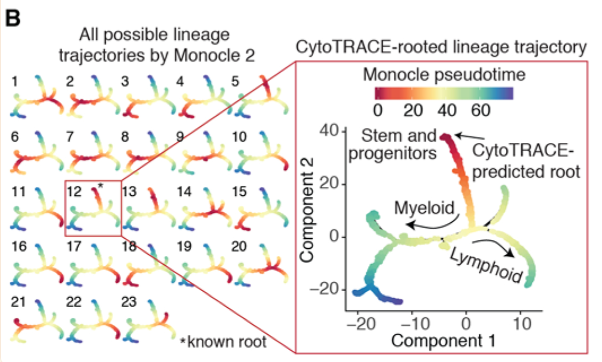
参考资料:
[1]Mapping single-cell developmental potential in health and disease with interpretable deep learning
[2]Single-cell transcriptional diversity is a hallmark of developmental potential
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-04-25,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号