透过蛋白质语言模型重新思考分子进化

透过蛋白质语言模型重新思考分子进化

DrugIntel
发布于 2026-06-24 14:10:15
发布于 2026-06-24 14:10:15

文献来源 Fernández R, Valverde S, Bombarely A, Cases I, Handley SA, Rojas AM. Rethinking molecular evolution through protein language model embeddings.Trends in Genetics (Forum), 2026. DOI: 10.1016/j.tig.2026.05.014
摘要
这是一篇立场鲜明的观点文章。作者主张:蛋白质语言模型(protein language models, pLMs)将序列压缩成的高维嵌入(embeddings),为分子进化提供了一种全新的、基于几何而非比对的研究视角——同源、分化、趋同等经典概念,可以被重新表述为高维表示空间中的拓扑与几何问题。但全文的真正分量,不在于鼓吹工具,而在于一个克制而深刻的认识论判断:pLM 学到的是进化的结果(自然选择与漂变在序列空间中留下的生化、结构与功能约束),而非进化的过程本身——它没有任何机制去追踪突变的时序、分歧的时间或谱系的亲缘关系。基于此,作者呼吁用分子进化与定量遗传学的完整工具箱,去"审讯"这些被模型默默总结好、却尚未被我们读懂的嵌入空间,并提出了一份具体的研究议程,以及一条"不取代、而是综合"的整合路线。
一句话:AI 已经替我们把蛋白质进化总结成了一张高维地图;现在的任务,是学会读懂这张地图的坐标。
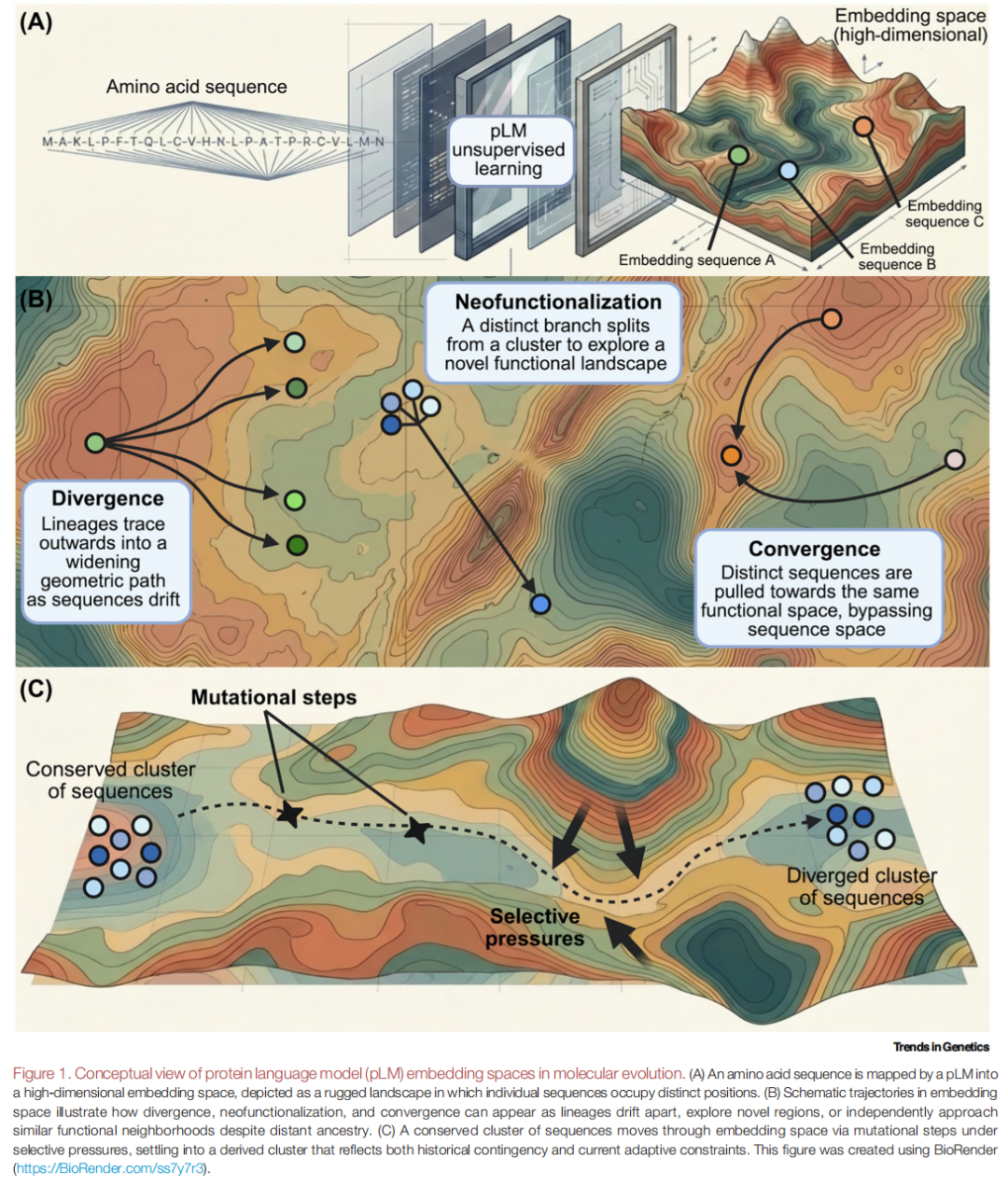
一、为什么需要重新思考?——经典范式及其结构性困境
分子进化的方法论根基,是把同源性(homology)作为一个关于共同祖先的可检验假设,通过序列比对(pairwise / multiple alignment)来推断。这条主线从 Dayhoff 的突变矩阵,一路延伸到现代基于最大似然和贝叶斯方法的系统发育学:它依赖明确的替换模型和有理论支撑的汇总统计量,把序列分歧定量地翻译为时间、结构与功能上的距离。
这一框架取得了巨大成功,却也在两类熟悉的情形下捉襟见肘:
- • twilight zone:当序列同一性低到某个临界点,同源信号被噪声淹没,比对方法难以区分"真正的远缘同源"与"趋同或偶然的相似"。
- • 功能未知序列的洪流:基因组与宏基因组测序产出了海量蛋白,其中相当大一部分功能完全未被注释,即所谓"暗蛋白质组"(dark proteome)。面对这种规模,逐条比对在通量与灵敏度上都难以为继。
更深一层的问题在于经典参数模型所依赖的简化假设。传统替换模型通常假定位点独立(site independence)与上下文无关的替换过程(context-free substitution),即一个位点如何演化,与序列其余部分无关。这些假设让模型在数学上可解、在计算上可行,却系统性地忽略了蛋白演化中普遍存在的高阶依赖——位点之间的上位效应(epistasis)、结构域层面的约束等。
需要强调的是,作者并未把多序列比对(MSA)一笔勾销。MSA 在计算序列谱、构建进化模型、辅助三维结构预测等任务上依然不可替代。问题不是"要不要 MSA",而是"经典框架是否已经触及了它能表达的进化信息的上限"。
二、蛋白质语言模型与嵌入空间:把序列"压缩"成一个坐标
pLM 提供了一条与比对范式正交的技术路线。
训练范式。这类模型通常在数亿条天然蛋白序列上以自监督方式训练,学习目标各异——掩码语言建模(masked language modeling,如编码器式架构)、自回归(autoregressive,如生成式架构)或对比学习(contrastive)。它们学的不是人类语言,而是氨基酸序列内部的统计规律。
嵌入的本质。训练完成后,模型可将任意序列(或每个残基)映射为一个高维数值向量,维度通常从数百到数千不等。这个向量——即嵌入——在无任何显式监督的情况下,编码了该蛋白的生化性质、二级与三级结构、功能角色,乃至进化约束。值得注意的是,正因为嵌入能捕捉序列内部的高阶相互作用,它也包含了传统位点独立模型在原理上无法表达的信息;这既是其威力所在,也意味着它与经典工具之间存在根本的表达能力落差。
视角的转变。一旦每条蛋白都成为高维空间中的一个点,那些历史上被框定为"同源搜索"或"家族分类"的任务,就自然转化为几何与拓扑问题:聚类、近邻、流形、轨迹。作者用一个贯穿全文的隐喻来描绘这个空间——一片崎岖的景观(a rugged landscape),其高低起伏由结构、功能与进化的约束共同塑造,而这一切都是从序列数据中"学"出来的。在这个意义上,pLM 揭示的是蛋白演化的系统性特征,而不只是序列层面的字母模式。
结构作为旁路。除了直接使用嵌入,还有一条互补路线是把 pLM 当作结构预测器。诸如 ESMFold 之类的工具,能在不依赖 MSA 的前提下,以基因组规模生成原子分辨率的结构模型。这为一类此前受限于"必须有实验解析结构"的分析——例如结构系统发育学(structural phylogenetics)——打开了通量化的大门。由于结构比序列更保守,这条路线尤其有望延伸到序列信号早已饱和的深层演化尺度。
几何直觉:在这片景观上会出现两类反直觉的现象——(1) 两条已分化到比对完全失效的序列,可能因折叠或功能相似而在嵌入空间中彼此邻近;(2) 两条亲缘很近的序列,可能因功能特化而落入相距甚远的区域。嵌入空间的"近",并不简单地对应系统发育上的"近"。
三、全文的认识论内核:嵌入捕捉的是进化的结果,而非过程
如果只能记住这篇文章的一个论断,应当是这一个。
pLM 从天然序列中学到的,是进化的结果(consequences)——即自然选择与遗传漂变,在序列空间中留下的那些生化、结构与功能约束的"印记"。但它并不掌握进化的过程(process)本身:它没有任何内在机制去追踪突变发生的时序、物种分歧的时间,或谱系之间的亲缘拓扑。
这一区分为何关键?可以从统计学角度把它讲透:
- • pLM 看到的是"幸存者"的分布。训练语料是当下存活的序列,它们逼近的是受约束序列空间的某种稳态分布(stationary distribution)——即"什么样的序列是可行的"。而系统发育推断需要的是沿着一棵树的替换过程(一个在时间上展开的马尔可夫过程),即"序列是如何一步步变成今天这样的"。前者是空间的截面,后者是时间的轨迹;二者并不等价。
- • 嵌入相似度 ≠ 系统发育距离。正因如此,二者完全可能彼此背离,尤其在精细尺度上:有时哪怕单个氨基酸的改变,都可能让向量表示发生显著位移。把嵌入距离直接当作演化距离来用,在原理上是有风险的。
- • 嵌入是"构造",不是"数据"。这是另一个常被忽视、却极其重要的方法学要点:嵌入并非对自然的直接测量,而是从数据中构造出来的产物,其几何形状被模型架构、训练目标与训练集组成三者共同塑造。这意味着,对嵌入距离做统计推断时,我们处理的并不是中性的"观测值",而是携带模型印记的人工对象。
作者由此给出了那个广为引用的判断:pLM 不只是会做预测的黑箱,它们是已经替我们把蛋白质进化总结好、而我们尚未读懂那份总结的黑箱。承认"嵌入是进化的结果而非过程"这一边界,正是能否严谨地将其用于进化推断的前提。
四、把进化视作"运动":嵌入空间中的轨迹与坐标系
在上述认识论约束之内,作者提出了一个富有生产力的重构:进化即运动——谱系在嵌入景观中移动,留下可被研究的轨迹。三类经典进化情形,在这个框架下获得了几何表述:
- • 分化(divergence):序列持续积累突变,谱系如扇形般向外铺展,在空间中走出越来越宽的路径。
- • 新功能化(neofunctionalization):基因复制之后,某一拷贝从祖先簇中剥离出去,像一条支线探入此前未被占据的功能区域。
- • 趋同(convergence):两条本无亲缘关系的序列,因承担相似功能而在空间中不约而同地弯向同一区域——绕过序列层面的相似,直接在功能空间相遇。
这种"运动"视角已经被进化速度(evolutionary velocity)框架部分实现(Hie et al., 2022)。该方法利用 pLM 对局部突变偏好的预测(本质上是模型对各种单点突变的相对偏好打分),在嵌入空间中构造出一个蛋白演化的向量场,从而为序列指定方向性与先后次序。它已被证明能在从病毒免疫逃逸到真核蛋白家族多样化的不同时间尺度上,还原出有方向的演化轨迹。
对进化生物学家而言,一个有用的心智模型是:嵌入把序列定位在一个习得的、上下文相关的隐空间(learned, context-dependent latent space)中。一旦我们能定义合适的定量坐标轴与参考点,这个隐空间有望被转化为一个显式的坐标系。与经典参数模型最根本的区别在于:模型不再假设单一的、上下文无关的替换过程,而是学到了一个突变的命运如何依赖于它所处的结构与功能环境。在序列同一性很低、比对失效的情形(例如许多内在无序蛋白)中,这种"上下文敏感"的优势尤为明显。
五、嵌入空间已经告诉了我们什么:证据链
把嵌入空间真正当作研究对象去做实验,已经积累了一批有意思、甚至带有挑战性的结果。它们既展示了潜力,也清醒地标出了边界:
(1) 直系同源猜想被打了个问号(Shaw et al., 2025)。 用嵌入去检验"一对一直系同源是否比旁系同源功能更保守"这一经典假设,结果是:在序列分歧程度可比的前提下,直系同源与旁系同源在嵌入空间中的相似度并无明显差异。这暗示嵌入空间并不能直截了当地区分"功能保守"与"功能分化",对长期被默认的 ortholog conjecture 构成了实证压力。
(2) 系统发育信号存在,但在富含插入/缺失的历史中系统性失真(Tule et al., 2024)。 嵌入能在一定程度上重现蛋白家族内部的系统发育关系,却在 indel 丰富的演化历史中出现系统性偏差。耐人寻味的是,这恰恰也是比对方法最容易出错的区域——于是问题变成:在这类困难区段,究竟是嵌入还是比对更接近真实的进化信号?目前悬而未决。
(3) 对趋同进化敏感,并揭示其复杂的序列基础(Cao et al., 2025)。 pLM 能把独立演化、但功能相似的序列在空间中拉到一起,从而揭示出趋同背后远比成对同一性所能展示的更为复杂的序列基础。
(4) 暗蛋白质组的规模化注释(Barrios-Núñez et al., 2024;Martínez-Redondo et al., 2025)。 基于嵌入相似度的方法(如 FANTASIA)已经能够在整个动物生命之树的尺度上,对功能未知的"暗蛋白质组"进行注释——这给出了用嵌入空间研究基因与基因组演化的一个切实可行的范例。
(5) 把 MSA 信息显式塞回 pLM,并不增益、甚至有害(Erckert & Rost, 2024)。 将 MSA 衍生的进化信息显式并入现代 pLM,在标准预测任务上一般不会带来提升,有时反而降低性能。这强有力地暗示:这些模型很可能已经隐式吸收了 MSA 所能提供的大部分信号。但作者随即提醒——这并不让经典工具变得多余(详见第八节)。
(6) "进化推理"在标准用法下并不会自动涌现(Ektefaie et al., 2025)。 有研究指出,在 pLM 的常规使用方式下,真正意义上的进化推理能力并未自发出现——这与"嵌入捕捉的是结果而非过程"的核心论断相互呼应,提醒我们不要把"相关性"误读为"机制"。
综合来看,证据呈现出一种审慎的乐观:嵌入确实承载了可观的进化相关信息,但它与经典量(同源、约束、分歧)的关系是有条件、依模型、依区段的,远非简单的等号。
六、黑箱的悖论与方法学挑战
嵌入空间的强大,恰恰伴随着一组尚未解决的难题。
坐标轴不可读。 我们能看到相似蛋白聚成簇、进化关系留下可辨认的图案,却很少知道空间中某个特定方向究竟对应哪一种生化、结构或功能变化。换言之,我们手握地图,却不认识图例。近期围绕稀疏自编码器(sparse autoencoders;Gujral et al., 2025)和 pLM 可解释性(Hunklinger & Ferruz, 2026)的工作,正是朝"读懂坐标轴"迈出的努力;另有研究表明 pLM 学到了相互作用序列基序的进化统计(Zhang et al., 2024),为"模型到底编码了什么"提供了线索。
模型依赖与普适性危机。 不同 pLM(掩码 vs 自回归 vs 对比;训练数据的分类学广度不同)会产生几何结构显著不同的嵌入空间。这带来一个尖锐的普适性问题:在某一模型嵌入空间中发现的进化规律,换一个架构可能就不成立。因此,跨模型比较是从任何单一模型得出稳健进化结论之前的必要前提。
采样偏差被几何化。 嵌入会继承现有序列数据库的盲点。已有工作指出 pLM 受物种代表性不均的训练数据所偏置(Sawhney et al., 2025;ProGen2,Nijkamp et al., 2023),而系统发育上的不平衡会直接扭曲我们想要研究的那片景观的几何形状。先识别并校正这些偏差,是任何系统性进化分析的前置条件——否则我们可能把数据库的伪影,误读为生物学的信号。
七、一份系统性研究议程(原文 Box 1)
作者并不止于提出概念,而是给出了一份相当具体的开放问题清单,勾勒出"系统性进化嵌入分析"作为一个新方向的核心议题:
# | 核心问题 | 要害 |
|---|---|---|
1 | 能否从嵌入中提取物种层面的进化信号? | 跨物种的嵌入距离,反映的是共同祖先,还是仅仅是蛋白家族内的结构/功能相似?在何种条件下能有意义地分离出物种级信号? |
2 | 嵌入与直系/旁系同源的关系如何? | 在大型、结构域重排的家族中,嵌入距离能否修正甚至推翻同源判断?何时它比同源或共线性更能预测功能保守? |
3 | 基因库演化在嵌入空间中是何形态? | 基因复制、丢失、水平转移、结构域重排,如何表现为轨迹或拓扑特征?能否在几何上把它们区分开? |
4 | 全蛋白质组嵌入能否作为可比较的"性状"? | 一个物种嵌入分布的汇总量,是否与生态、生活史、基因组结构或发育复杂度相关?能否用于系统发育基因组学推断? |
5 | 经典模型与嵌入在哪里分歧、为何分歧? | 在序列空间的哪些区域,显式进化模型与嵌入推断会就同源、约束或趋同给出冲突答案?我们能从这些分歧中学到什么? |
6 | 嵌入能否捕捉物种形成、多样化、适应等过程? | 其中是否藏有谱系分裂、多样化动态变化、适应性约束转移的可识别签名?如何用独立的系统发育与群体遗传基准来稳健验证? |
7 | 进化模式在不同 pLM 间有多普适? | 在某一架构/目标/层/数据集中检测到的进化信号,能在多大程度上推广到其他模型?模型间差异又揭示了哪些进化信息是稳健编码的、哪些只是训练选择的产物? |

这七问的共同指向是:把嵌入从"相似度搜索的辅助特征",提升为值得用进化与群体遗传方法严肃研究的对象。
八、面向参数的回归,与一场 新综合
三步研究纲领
作者把上述议程落地为一个具体的研究纲领:
- 1. 测绘景观(map the landscape):系统性地探查不同架构、训练目标与数据集下的嵌入空间,弄清哪种生化、结构与进化属性被编码在何处,核心目标是提升可解释性。
- 2. 研究演化路径(study evolutionary paths):把连接相关蛋白或蛋白质组的嵌入序列当作路径来分析,追问哪些突变类型与选择压力能够生成沿路径观察到的变化。
- 3. 回桥到参数(bridge back to parameters):把"一条序列在嵌入空间中近邻点的分布模式",翻译为上下文相关的突变发生与保留倾向,从而让嵌入去告知与精炼(而非取代)可解释的进化模型。
不是取代,而是双向奔赴
作者对 pLM 与经典方法关系的判断,凝练在一个精当的比喻里(此处转述其意):
一个只会用 pLM 的系统发育学家,就像一位只拿着一张极其精细、却毫无标注的卫星图的地理学家——画面丰富而连续,但若缺乏解读它的知识,你根本不知道任何一个景观特征意味着什么。
由此引出一种两个方向的整合:
- • 正向:嵌入不应只是默默给现有流程打下手,而可充当带语境意识的先验(context-aware priors)——重塑我们如何搭建进化模型,从"该考虑哪些突变是合理的",到"该重点探索序列空间的哪些区域"。
- • 逆向:传统工具也不应被降格为事后验证,而应主动去拷问 pLM 学到了什么——把正选择信号、位点特异速率变化、约束改变,投射回那片潜在景观,看看模型对进化的理解,在哪里与我们的理论一致、又在哪里发生偏离。
生成式设计:把"是否学到进化结构"变成可检验命题
这种双向交流自然延伸到生成式蛋白设计。诸如 ESM-C 之类的模型能产生远离任何天然同源物的功能性序列(Hayes et al., 2025,"用语言模型模拟五亿年的演化")。其深刻之处在于:如果嵌入真的编码了进化约束,那么这些模型生成的新蛋白就理应遵守这些约束——于是,生成式设计本身,就成了对"pLM 是否真正学到了进化结构"的一次实证检验,而非仅仅是工程应用。
两条互补策略
一个不容回避的事实是:标准 pLM 的训练目标(如掩码语言建模)并未显式优化进化信号。这意味着嵌入中残留的任何进化信息,都更像是一种涌现的副产品而非设计初衷——其中一些进化上极有价值的变异,也可能在此过程中被丢失或扭曲。据此,作者给出两条互补路径:
- • 策略一(本文主张):用进化工具去"审讯"现有 pLM,弄清它到底保留了多少进化信号、又藏在何处。
- • 策略二(长远更有力):设计训练目标本身就显式纳入进化结构的新一代 pLM——例如在系统发育上更均衡的数据集上训练、把位点特异进化速率作为辅助监督、或并入比较基因组学信号;在现有模型上做面向进化任务的微调,则是一条务实的中间路线。
两条策略并不互斥:正是对现有模型的探查,能告诉我们哪些进化信息被系统性地丢掉了,从而直接指导进化感知架构的设计。
九、批判性评述:这篇文章的位置、贡献与未尽之处
它的真正贡献,是一次"概念校准"而非工具推介。 在 pLM 应用论文层出不穷的当下,本文最有价值的动作是划清边界——明确"结果 vs 过程""构造 vs 数据""相关 vs 机制"这几组区分。这把许多模糊的乐观,收敛为可被检验的命题。对一篇 Forum 而言,提出正确的问题(尤其是 Box 1 的七问)本身就是贡献。
几处值得继续追问的张力:
- • "回桥到参数"是纲领,尚非方法。 把高维近邻结构翻译为上下文相关的替换倾向,这一步在概念上极具吸引力,但如何具体实现、如何与现有的位点特异速率模型对接、如何做统计检验,文中仍是蓝图。这恰恰是该领域未来最硬的骨头。
- • "嵌入是构造"这一点,对统计推断的冲击或被低估。 如果嵌入几何随架构而变,那么"嵌入距离的显著性"本身就缺乏一个稳定的零模型(null model)。在建立可跨模型迁移的、有统计保证的距离度量之前,许多基于嵌入的进化结论都应被视为探索性的。
- • 可解释性是整套愿景的咽喉。 "读懂坐标轴"不是锦上添花,而是把嵌入从"黑箱总结"变为"可用进化地图"的必要条件。稀疏自编码器等方向(Gujral et al.)能走多远,很大程度上决定了本文愿景的可行性上限。
- • 验证基准的循环风险。 当我们用系统发育/群体遗传基准去验证嵌入,又反过来想用嵌入去修正这些经典推断时,需要格外小心循环论证:用 A 验证 B,再用 B 推翻 A,逻辑上必须有独立的第三方证据(如实验功能数据、结构、深层化石/地质标定)来打破闭环。
与领域的衔接。 本文与结构系统发育学(借 ESMFold 等把分析推向深层演化)、生成式蛋白设计(把约束的"被遵守"作为检验)、以及机制可解释性研究构成了一个自洽的研究网络。它没有宣称范式被推翻,而是更稳妥地主张:经典分子进化的解释力与假设检验框架,是读懂 pLM 这片"学习得到的景观"所不可或缺的罗盘。
结论性判断。 pLM 给了我们一幅比任何单一速率矩阵都更丰富的进化图景,但它不是显式模型与审慎推断的替代品。这篇文章的价值,正在于把"惊叹与不安"转化为一份可执行的研究日程:不是站在这片学习得到的景观前手足无措,而是发展出探索、测绘并解读它的概念与方法工具,并把所得洞见,与半个世纪以来积累的分子进化知识整合起来。理解蛋白如何演化的旅程并未结束——它只是有了一支新的罗盘。
关键术语速查
术语 | 含义 |
|---|---|
蛋白质语言模型(pLM) | 在海量天然蛋白序列上自监督训练的神经网络,能把序列映射为高维向量。 |
嵌入(embedding) | pLM 为序列/残基生成的高维数值向量,隐式编码生化、结构、功能与进化约束。 |
掩码语言建模(MLM) | 一种自监督目标:遮住部分残基让模型预测,典型于编码器式 pLM。 |
黄昏地带(twilight zone) | 序列同一性过低、同源信号难以从噪声中分辨的区域。 |
暗蛋白质组(dark proteome) | 功能未被注释的大量蛋白序列。 |
多序列比对(MSA) | 把同源序列对齐以提取保守/共变信号的经典方法。 |
上位效应(epistasis) | 不同位点之间相互影响其突变效应的高阶依赖。 |
直系/旁系同源(ortholog / paralog) | 因物种分化 / 因基因复制而产生的同源关系。 |
新功能化(neofunctionalization) | 基因复制后某拷贝获得新功能。 |
进化速度(evolutionary velocity) | 利用 pLM 的局部突变偏好,在嵌入空间构造演化方向向量场的框架。 |
结构系统发育学(structural phylogenetics) | 基于(预测)结构而非序列推断演化关系,适用于深层分歧。 |
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-21,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号