AI+Drug 文献速递 | 虚拟细胞:AI 驱动的药物发现新范式

AI+Drug 文献速递 | 虚拟细胞:AI 驱动的药物发现新范式

MindDance
发布于 2026-01-08 12:47:39
发布于 2026-01-08 12:47:39
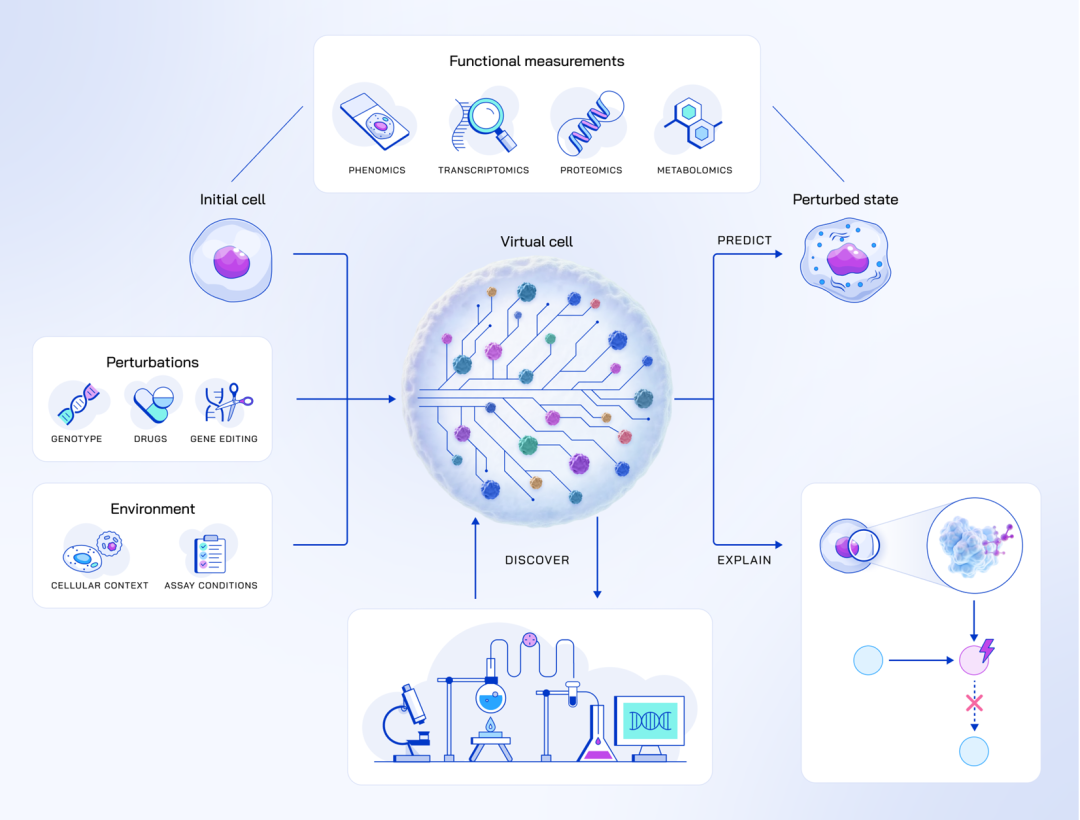
1. Virtual Cells: Predict, Explain, Discover
期刊: arXiv 链接: https://arxiv.org/abs/2505.14613
简介: 本文提出虚拟细胞(Virtual Cells)的愿景,旨在通过AI模型模拟细胞对扰动的功能响应、解释分子机制并发现新生物学见解。虚拟细胞需具备预测(如基因表达变化)、解释(如关键分子互作)和发现(如通过实验闭环生成假设)能力,结合高通量数据、AI/ML和自动化实验室技术构建。文中提出三大设计原则:基于细胞初始状态预测响应、动态建模分子互作、通过实验闭环迭代优化模型,并倡导生物学基准测试。在聚合物数据集等场景的实验中,虚拟细胞展现了对多模态数据的整合能力和跨尺度建模潜力,为药物发现提供了从分子到细胞层面的综合模拟框架,推动AI在精准医疗和靶点发现中的应用。
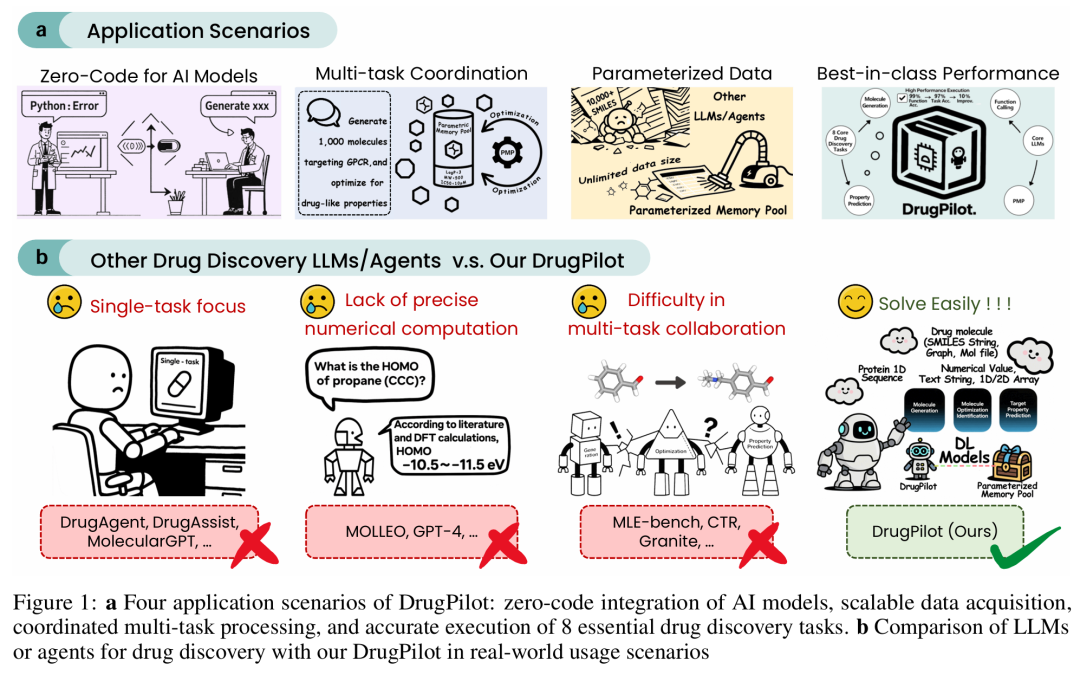
2. DrugPilot: LLM-Based Parameterized Reasoning Agent for Drug Discovery
期刊: arXiv 链接: https://arxiv.org/abs/2505.13940 代码: https://github.com/wzn99/DrugPilot
简介:本文提出DrugPilot,一种基于大语言模型(LLM)的参数化推理代理,旨在解决药物发现中多模态数据处理、领域知识更新延迟及复杂任务预测置信度不足的挑战。其核心通过参数化推理架构和交互式参数化内存池(PMP),将多模态药物数据标准化为参数表示,结合反馈与聚焦(Fe-Fo)机制纠正推理错误并维持任务焦点。实验基于自建的药物工具调用数据集TCDD(含8类任务、2800样本),在简单、多任务和多轮任务上分别实现98.0%、93.5%和64.0%的任务完成率,显著优于ReAct等基线模型。研究表明,DrugPilot通过自动化多阶段任务规划和执行,提升了药物发现的效率与准确性,为AI驱动的药物研发提供了新框架。
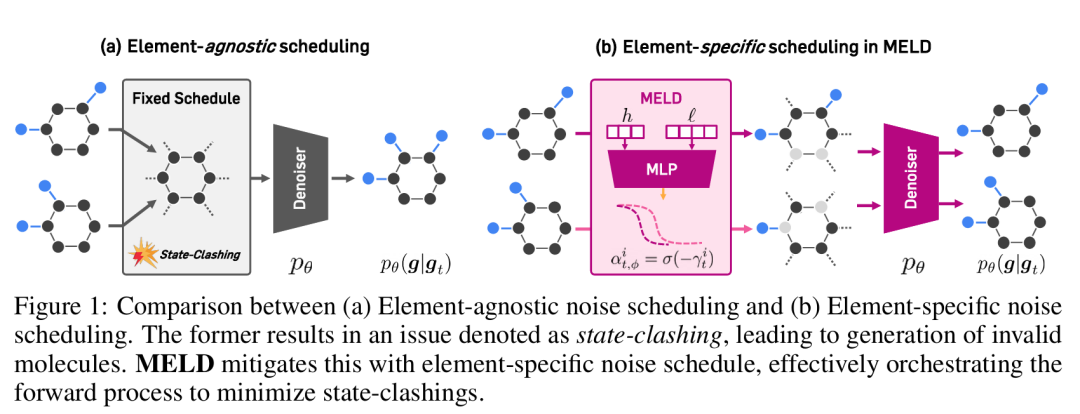
3. Learning Flexible Forward Trajectories for Masked Molecular Diffusion
期刊: arXiv 链接: https://arxiv.org/abs/2505.16790
简介: 本文针对掩蔽扩散模型(MDM)在分子生成中因元素无关噪声调度导致的“状态冲突”问题,提出Masked Element-wise Learnable Diffusion(MELD)框架。通过引入参数化噪声调度网络,为分子图的原子和键分配独立的腐化轨迹,避免不同分子在扩散过程中坍缩为相同中间状态。实验在QM9、ZINC250K和聚合物数据集上显示,MELD将香草MDM的化学有效性从15%提升至93%,在条件生成任务中属性对齐误差降低16.5%,显著优于基线模型。研究表明,元素级可学习扩散轨迹有效缓解了状态冲突,提升了分子生成的化学有效性和多样性,为离散分子图的生成提供了更优的扩散模型解决方案。
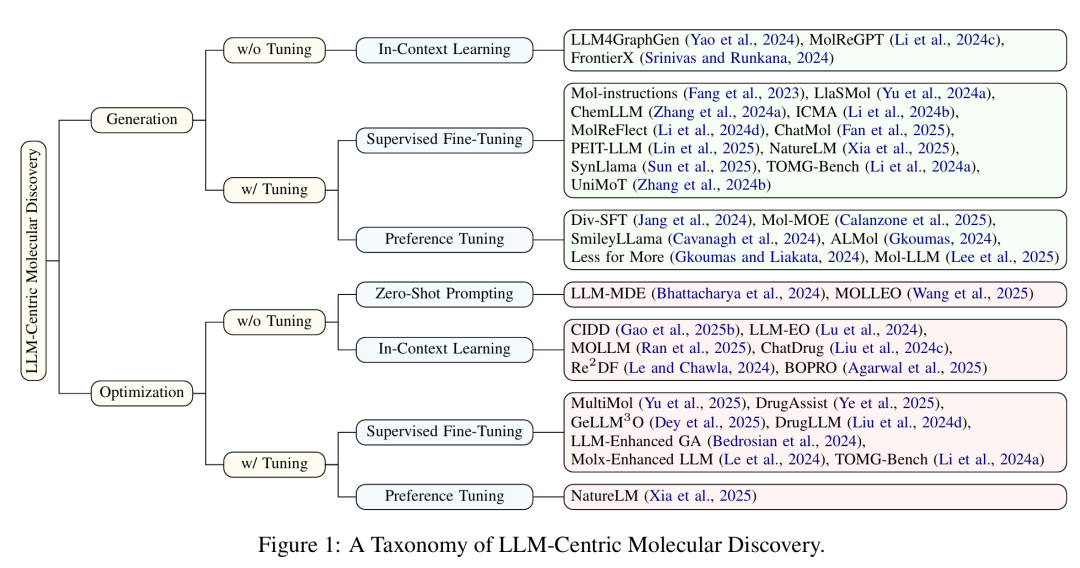
4. A Survey Of Large Language Models For Text-Guided Molecular Discovery: From Molecule Generation To Optimization
期刊: arXiv 链接: https://arxiv.org/abs/2505.16094v1 代码: https://github.com/REAL-Lab-NU/Awesome-LLM-Centric-Molecular-Discovery
简介: 该综述全面总结了大语言模型(LLMs)在分子生成与优化任务中的应用,揭示了其在化学空间中通过自然语言指导分子设计的潜力和挑战。文章提出了一种以学习范式为核心的全新分类方法,涵盖了无调优(如零样本提示、上下文学习)和有调优(如监督微调、偏好调优)两大类,并详尽回顾了代表性方法及其在化学任务中的适应性。论文还系统整理了常用的数据集与评估协议,并通过任务实例分析了LLMs如何理解和操控复杂的分子结构信息。在分析现有研究的基础上,作者指出当前LLMs在结构理解、属性控制和多模态融合方面的关键挑战,并展望了未来发展方向。该论文为从事分子科学与生成模型交叉研究的学者提供了重要资源与研究框架。
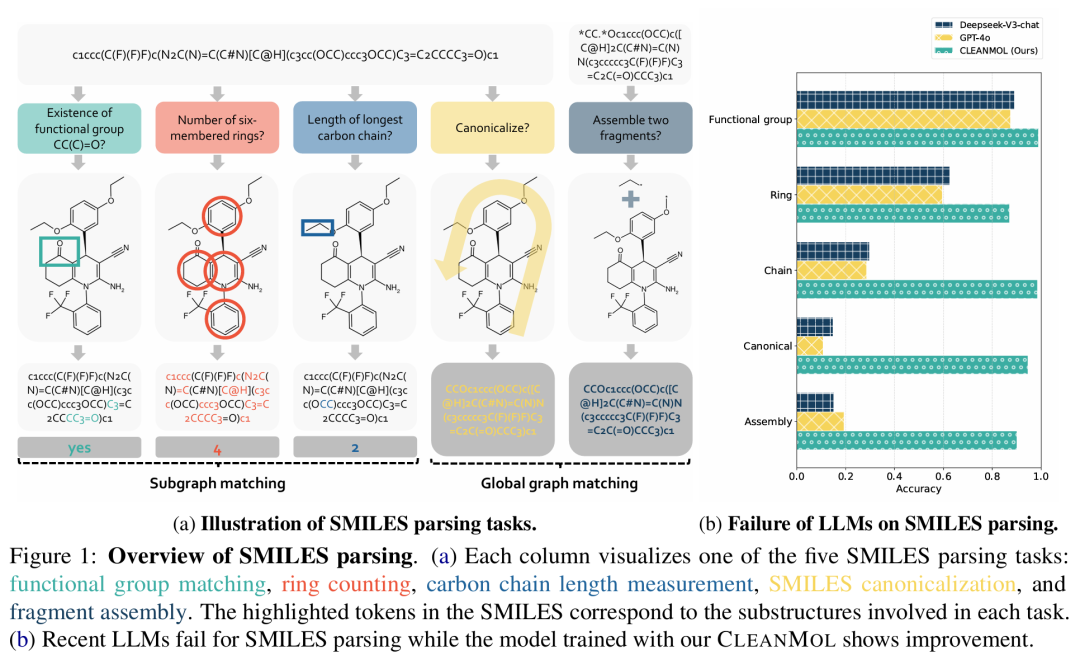
5. Improving Chemical Understanding Of LLMs Via SMILES Parsing
期刊: arXiv 链接: https://arxiv.org/abs/2505.16340v1
简介: 本文提出CLEANMOL框架,以提升大语言模型对SMILES分子表示的结构理解能力,解决其在基本结构识别任务上的表现不佳问题。作者设计了五类确定性、可扩展的SMILES解析任务,包括官能团识别、环计数、碳链长度测量、规范化和片段组装,全面覆盖局部与全局图结构信息。CLEANMOL通过构建自动标注的预训练数据集,并引入任务自适应数据筛选与课程学习策略,有效提升了模型的结构理解能力。在Mol-Instructions基准测试中的三项下游任务上,模型实现了领先或竞争力的表现,验证了结构感知预训练的有效性。该研究为LLMs在化学结构处理方面的基础能力构建提供了新的路径。
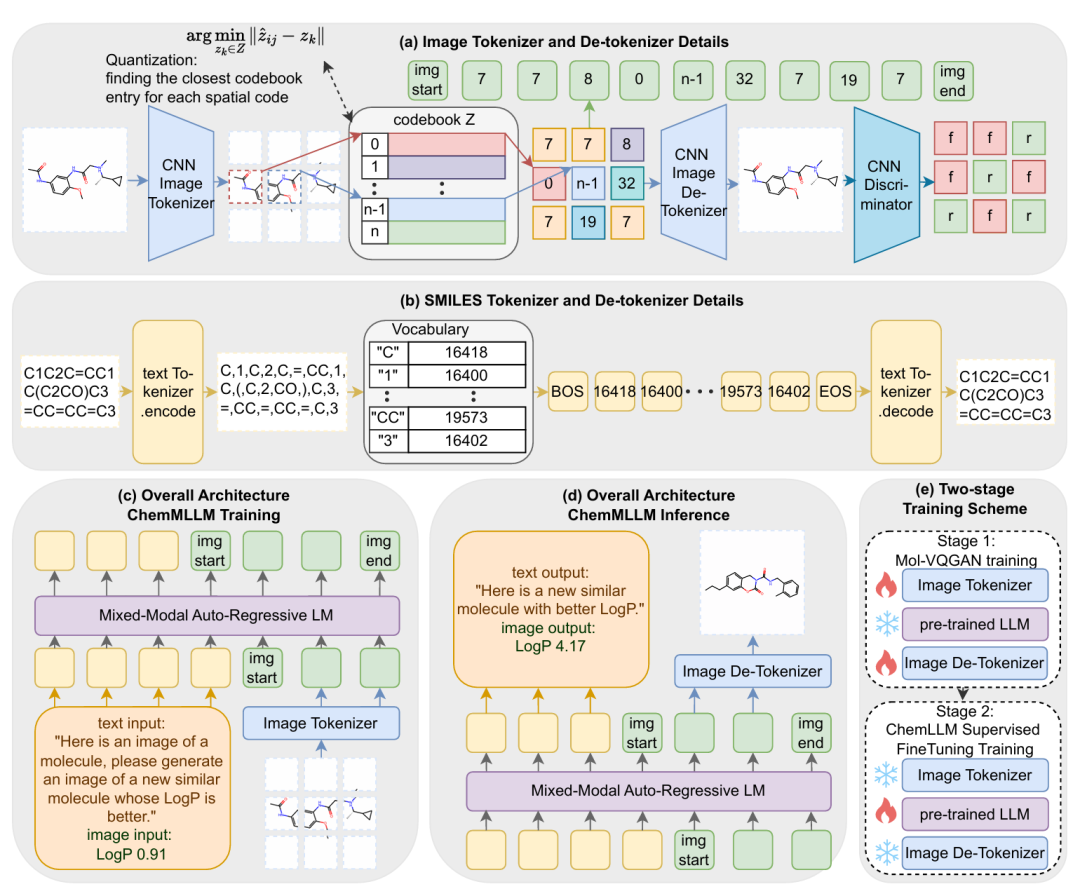
6. ChemMLLM: Chemical Multimodal Large Language Model
期刊: arXiv 链接: https://arxiv.org/abs/2505.16326v1 代码: https://github.com/bbsbz/ChemMLLM.git
简介: 本文提出ChemMLLM,首个统一支持文本、SMILES和分子图像三模态输入与生成的化学多模态大语言模型,旨在弥合当前模型在化学任务中感知与表达能力的不足。模型采用“图像分词器–LLM–图像解码器”的架构,并引入Mol-VQGAN以高效编码稀疏分子图像信息。在五项设计的跨模态任务上,ChemMLLM在图像生成、性质预测、描述生成等任务中显著优于现有通用多模态与化学LLMs,例如在分子图像优化任务中,其LogP提升幅度为GPT-4o的两倍以上。该模型展示了在统一框架下实现分子多模态理解与生成的潜力,为化学智能系统发展开辟了新路径。
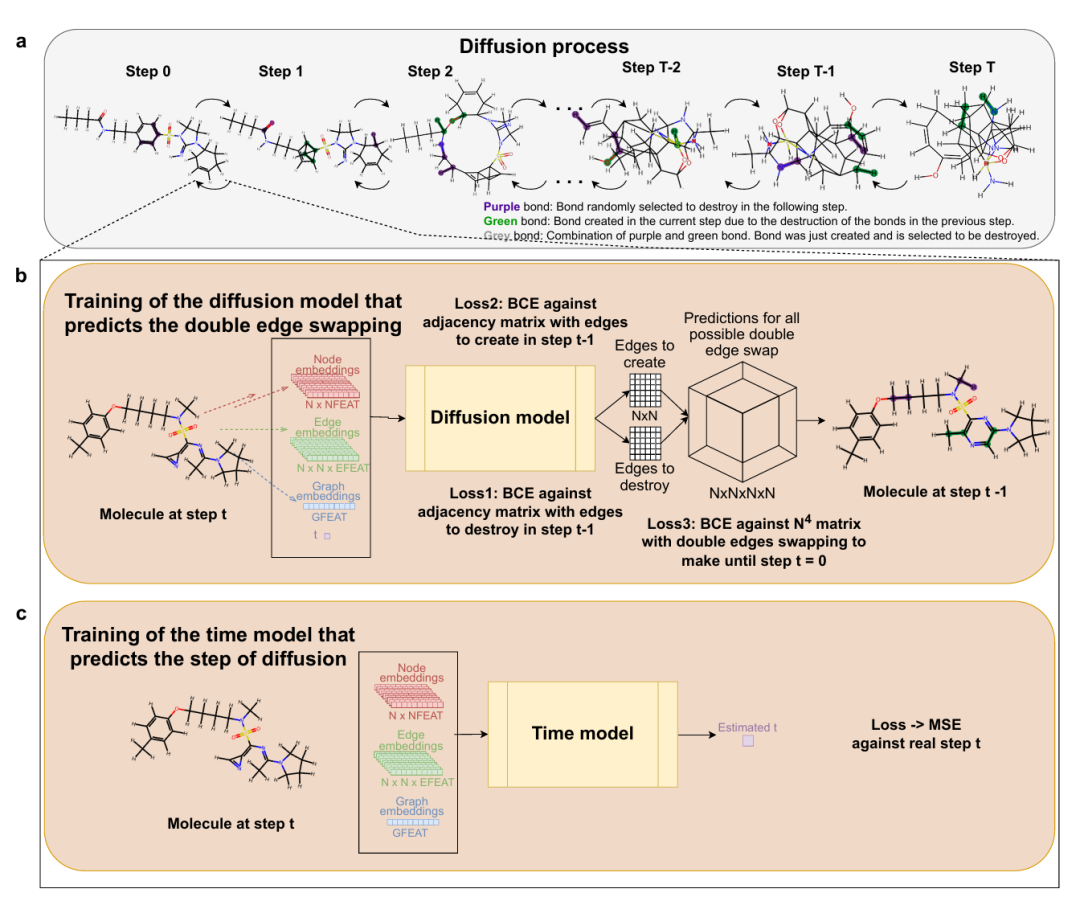
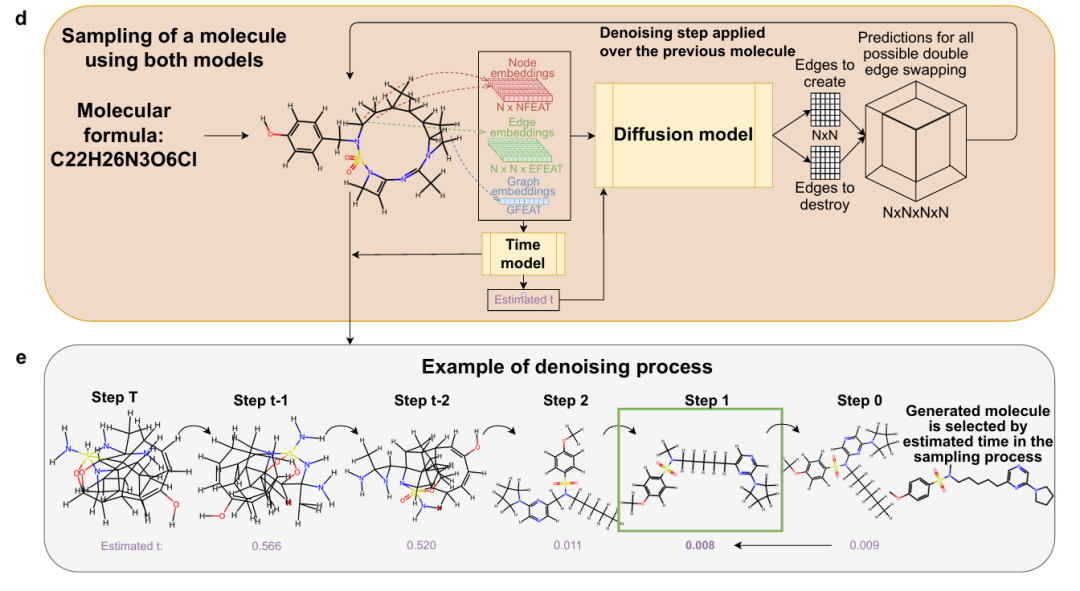
7. A Collaborative Constrained Graph Diffusion Model For The Generation Of Realistic Synthetic Molecules
期刊: arXiv 链接: https://arxiv.org/abs/2505.16365v1
简介: 本论文提出CoCoGraph,一种协同约束图扩散模型,可生成100%化学有效的新型合成分子。该方法采用双边交换机制,在扩散过程中强制维持原子化合价,从而确保分子的化学有效性,并通过协同机制联合训练两个模型:一个预测图结构变化,另一个预测当前扩散时间步,从而引导模型更有效地还原目标分子。实验采用GuacaMol基准对比JTVAE与DiGress等方法,在有效性、唯一性、新颖性和分子性质分布等指标上均实现最优表现,尤其在36种化学性质分布上表现出更接近真实分子的统计特征。基于模型的高效性,作者生成了820万个新分子,并开展了“图灵测试”验证其真实感,结果显示有机化学专业人士辨别生成分子与真实分子的准确率仅略高于随机猜测,进一步验证了模型的生成质量。该研究证明了将化学约束嵌入生成过程而非学习参数中的优势,提升了模型效率与生成质量,为新分子的设计和化学空间探索提供了强有力工具。
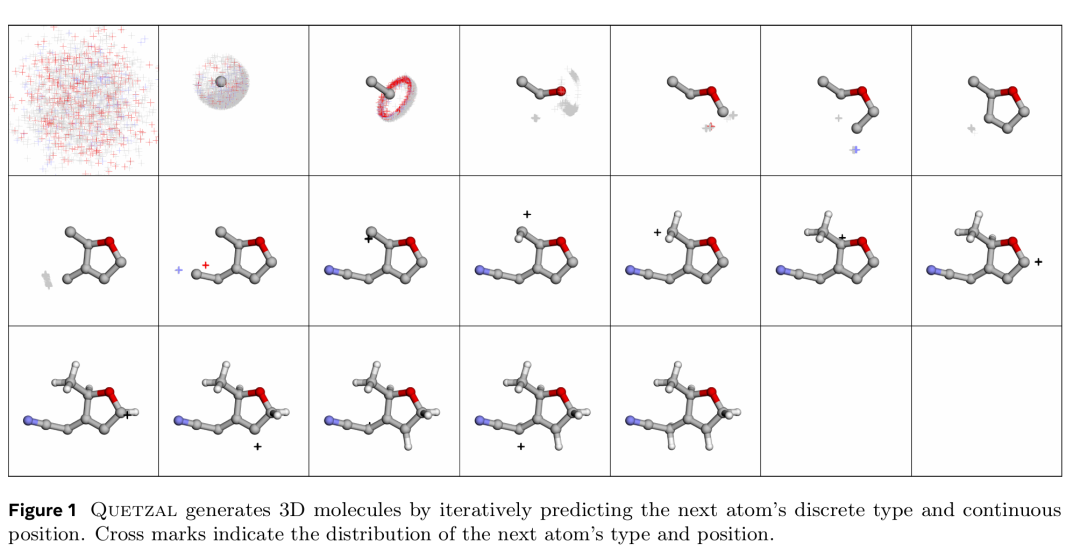
8. Scalable Autoregressive 3D Molecule Generation
期刊: arXiv 链接: https://arxiv.org/abs/2505.13791
简介: 本文提出Quetzal,一种可扩展的自回归模型,用于3D分子生成,解决了扩散模型计算密集和自回归模型生成质量不足的问题。Quetzal结合因果Transformer预测原子类型,使用扩散MLP建模连续坐标,实现了生成质量与速度的平衡。实验在QM9和GEOM数据集上进行,结果显示Quetzal在有效性和唯一性上超越现有自回归基线,接近最先进扩散模型性能,且生成速度显著更快,同时支持氢装饰和骨架补全等可变尺寸任务。该研究为3D分子生成提供了兼具高效性与泛化性的新范式,推动了分子生成模型在化学和材料科学中的应用。
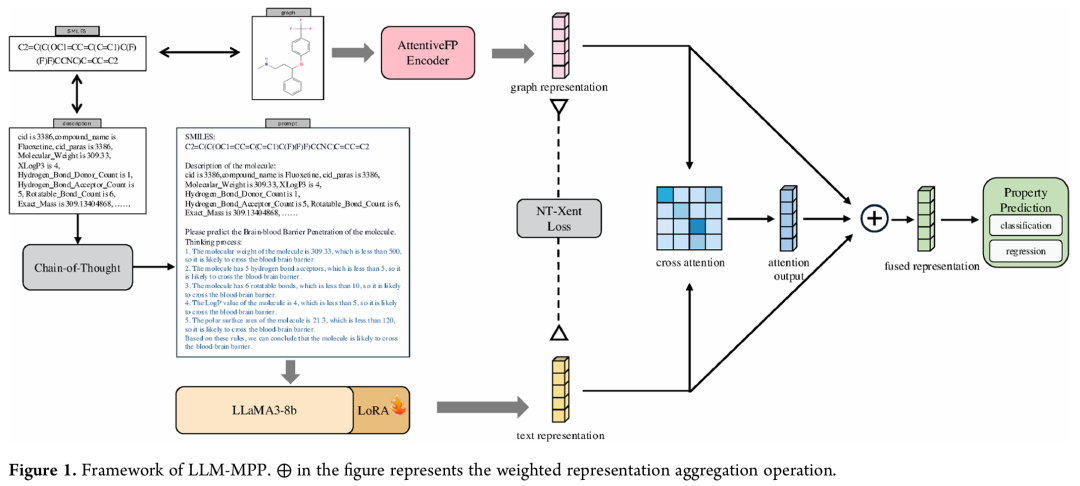
9. Effective and Explainable Molecular Property Prediction by Chain-of-Thought Enabled Large Language Models and Multi-Modal Molecular Information Fusion
期刊: Journal of Chemical Information and Modeling 链接: https://doi.org/10.1021/acs.jcim.5c00577 代码: https://github.com/jinchang1223/LLM-MPP
简介: 本文提出LLM-MPP,一种基于大语言模型(LLM)的多模态分子性质预测方法,融合1D SMILES、2D分子图和文本描述,通过思维链(CoT)技术增强可解释性,利用交叉注意力和对比学习实现多模态特征融合。在MoleculeNet的9个基准数据集上,LLM-MPP在5个数据集上表现最优,2个排名第二,超越22个基线模型。消融实验验证了各模块有效性,t-SNE可视化显示模型能有效区分分子表示,案例研究成功预测真实药物的血脑屏障穿透性和毒性。该方法为分子性质预测提供了可解释的多模态解决方案,提升了药物发现的效率和可靠性。
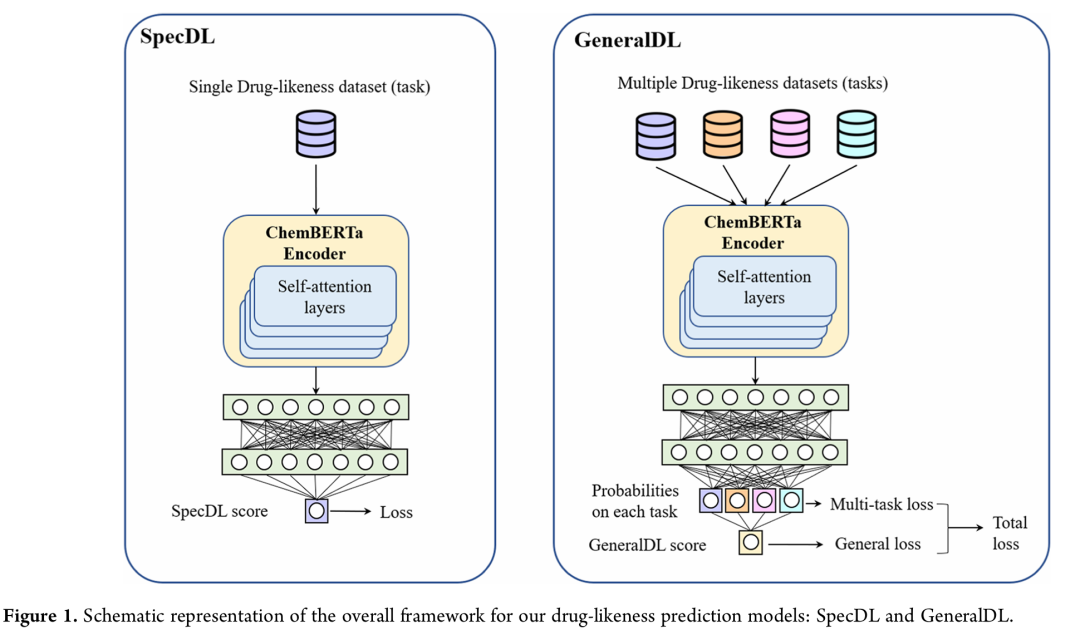
10. Comprehensive Drug-Likeness Prediction Using a Pretrained Transformer Model and Multitask Learning
期刊: Journal of Chemical Information and Modeling 链接: https://doi.org/10.1021/acs.jcim.5c00455 代码: https://github.com/EricCai00/druglikeness
简介: 本文开发了结合预训练Transformer和多任务学习的药物相似性预测框架,包含专用模型SpecDL和通用模型GeneralDL。SpecDL在4个任务上平均ROC-AUC达0.836,GeneralDL在6个内外测试集上平均0.781,均优于现有基线。实验利用多源数据集(如Approved + ZINC、Clinical Milestone)验证模型泛化能力,发现GeneralDL在毒性和生物活性预测中表现稳健,注意力权重分析揭示其可解释性。研究构建的网络服务器提供了便捷的药物相似性评估工具,为早期药物筛选提供了高效、可解释的计算框架,助力降低临床试验失败率。
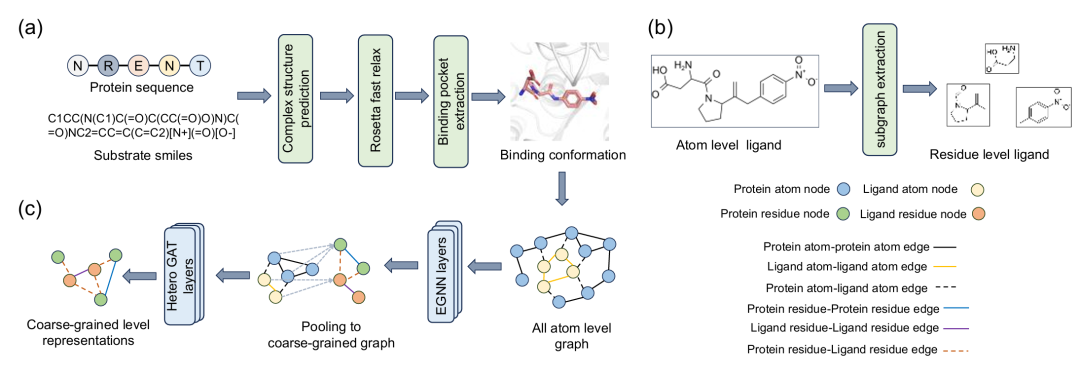
11. Catalytic pocket-informed augmentation of enzyme kinetic parameters prediction via hierarchical graph learning
期刊: bioRxiv 链接: https://doi.org/10.1101/2025.05.18.654694 代码: https://github.com/DingLuoXMU/GraphKcat
简介: 本文提出GraphKcat框架,通过整合酶-底物三维结合构象和多模态特征,提升酶动力学参数(Kcat/Km)预测的准确性和泛化性。模型利用Chai-1生成复合物构象,构建从全原子到粗粒度的多尺度图神经网络(GNN)提取活性位点特征,并通过多模态交叉注意力融合模块(MMCAF)与LLMs编码的序列、底物和环境信息结合。实验在MPEK数据集上显示,GraphKcat在低序列相似性条件下(<40%)的Kcat预测PCC达0.432,超越纯语言模型基线5.6%,且对结构质量不敏感,能准确识别高活性突变体。该研究为酶工程和挖掘提供了结合结构与序列信息的有效工具,尤其在低数据场景下展现出优势。
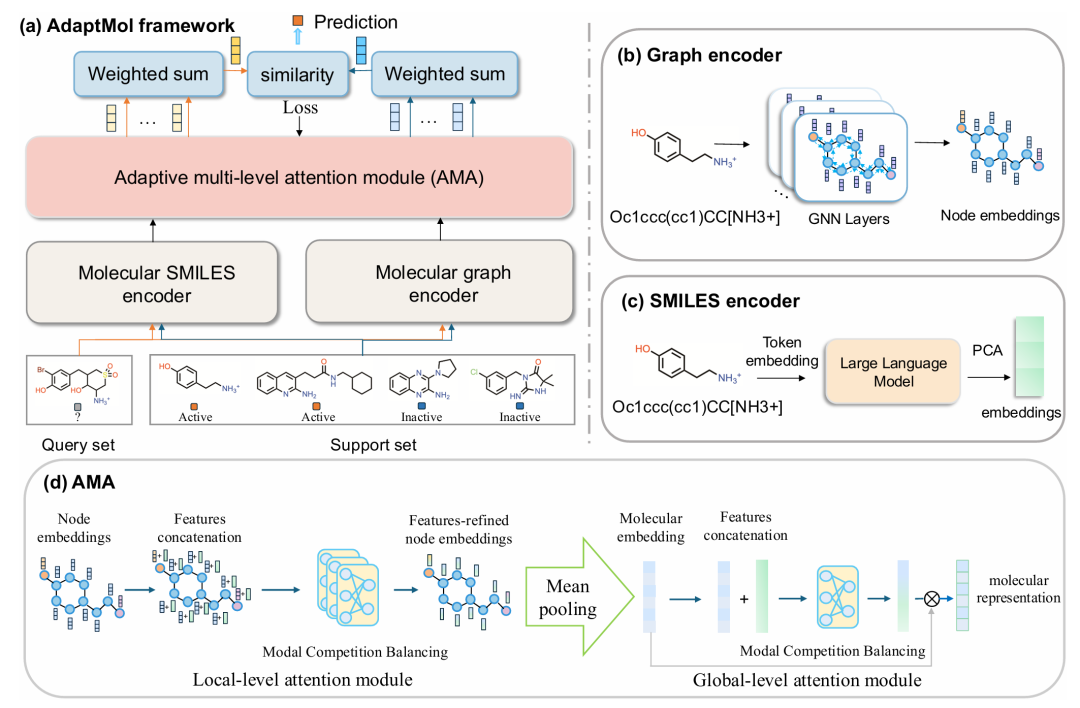
12. AdaptMol: Adaptive Fusion from Sequence String to Topological Structure for Few-shot Drug Discovery
期刊: arXiv 链接: https://arxiv.org/abs/2505.11878v1
简介: 针对小样本药物发现中分子表示不足的问题,AdaptMol提出自适应多水平注意力模块(AMA),动态融合SMILES序列的全局语义与分子图的局部拓扑特征。模型通过图编码器提取原子相互作用和子结构信息,结合LLMs生成的序列特征,在5-shot和10-shot设置下对Tox21、SIDER等基准数据集进行评估,ROC-AUC平均优于现有基线2.44%-4.18%。解释性分析显示,AMA能识别关键活性子结构,如BACE抑制剂中的酰胺键和胺基,验证了多模态融合的有效性。该框架为小样本场景下的分子性质预测提供了可解释的解决方案,提升了模型在化学空间中的泛化能力。
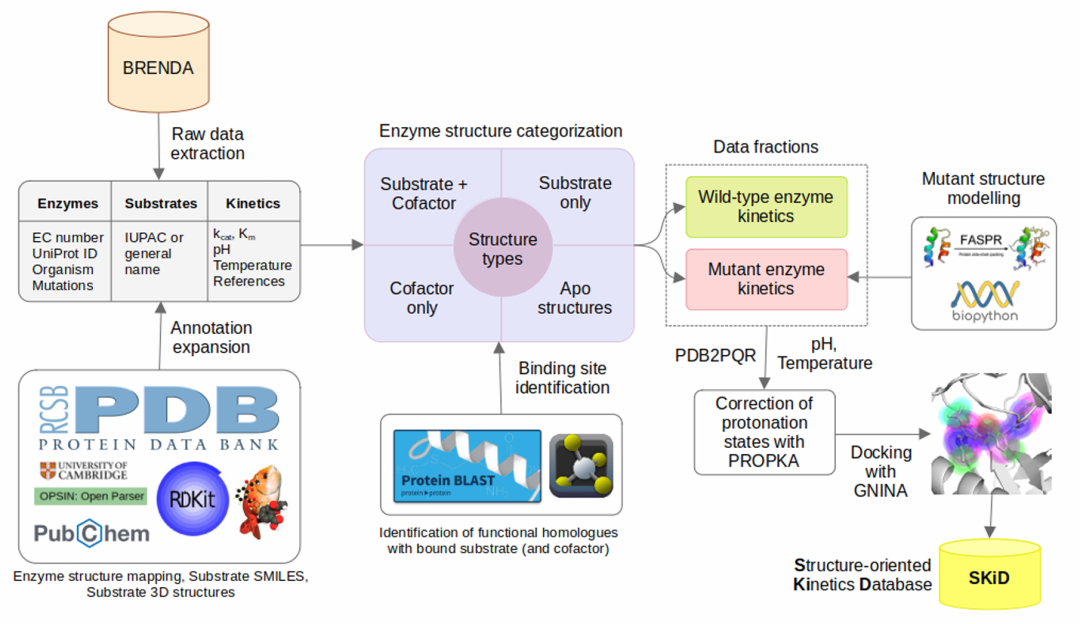
13. SKiD: A Structure-Oriented Kinetics Database of Enzyme-Substrate Interactions
期刊: bioRxiv 链接: https://www.biorxiv.org/content/10.1101/2025.05.18.654770v1
简介: SKiD是首个整合酶-底物动力学参数(kcat/Km)与三维结构的数据库,通过BRENDA数据结合结构预测工具(如FASPR、GNINA)构建,包含13,654个独特复合物,覆盖6类酶。数据经质子化状态校正、突变映射和分子对接等处理,解决了结构-功能关联的缺失问题。数据库收录野生型和突变体酶的实验数据,支持结合位点注释和环境条件(pH、温度)记录。技术验证显示,GNINA对接在FASPR建模的突变体结构上表现接近实验数据,RMSD差异仅0.71Å。该资源为酶工程、代谢模型构建及AI预测模型训练提供了关键数据支撑,推动结构-动力学关联研究。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-05-24,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号