J. Med. Chem. | 重磅综述告诉你:口服药,长啥样?

J. Med. Chem. | 重磅综述告诉你:口服药,长啥样?

DrugOne
发布于 2025-11-29 17:23:03
发布于 2025-11-29 17:23:03
在现代药物研发中,“一日一次、低剂量、高安全性”常被视为口服药物设计的理想目标。
然而,Jnana Therapeutics的Dean G Brown近期发表于《药物化学杂志》的综述文章通过对2020至2024年间美国FDA批准的104种小分子口服药物进行系统性分析,揭示出现实中药物的真实面貌远比我们想象中更加多样与复杂。
该研究从剂量方案、药代动力学特性、安全性特征、代谢行为及化学结构等多个维度展开剖析,为药物研发者提供了超越传统教条的宝贵视角与数据支持。

剂量方案:并非所有药物都一日一次

在剂量方案方面,尽管一日一次(QD)仍是主流,占所分析药物的67%,但仍有28%的药物采用一日两次(BID)给药,4%采用一日三次(TID),甚至有一种药物托沃拉非尼(Tovorafenib)采用每周一次的方案。
这一数据清晰地表明,并非所有成功的口服药物都必须满足QD给药的苛刻标准。
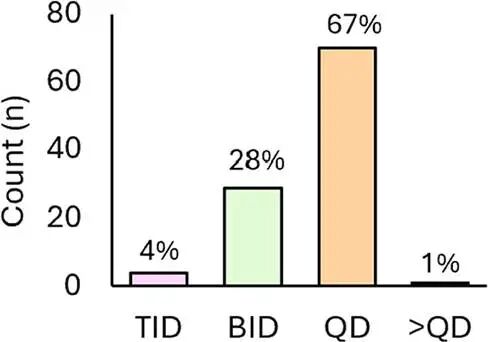
2020-2024FDA批准获批药物剂量方案统计
进一步分析发现,首创药物(FIC)与孤儿药(Orphan)中采用BID或TID方案的比例显著更高。具体而言,50%的首创药物采用BID/TID方案,而非首创药物中这一比例仅为19%;在孤儿药中,BID/TID比例为41%,而非孤儿药中仅为20%。
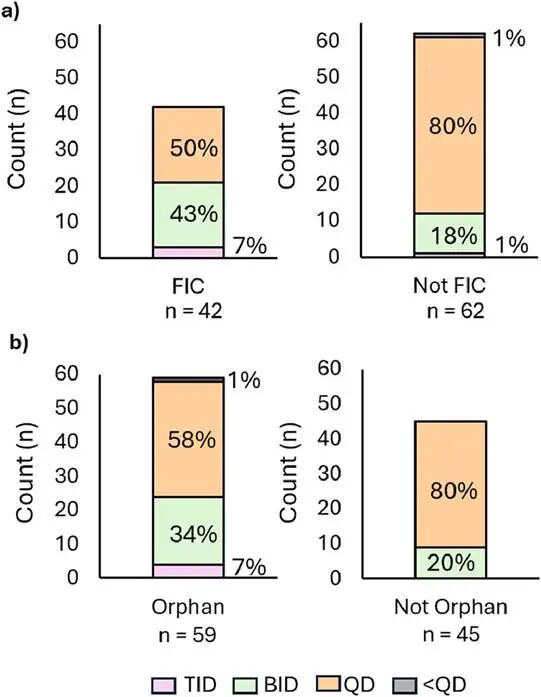
获批首创药物或孤儿药剂量方案统计
这一现象反映出在治疗需求迫切、患者群体有限或机制全新的疾病领域中,临床更倾向于接受更频繁的给药方案以换取疗效的突破。
剂量分析还显示,首创药物的中位日剂量为200毫克,显著高于非首创药物的122毫克;孤儿药的中位日剂量同样为200毫克,远高于非孤儿药的50毫克。
这些数据说明,在特定临床背景下,较高的剂量与较频繁的服药方式仍具有其合理性与可接受性。
药代动力学:高蛋白结合与高清除率并存
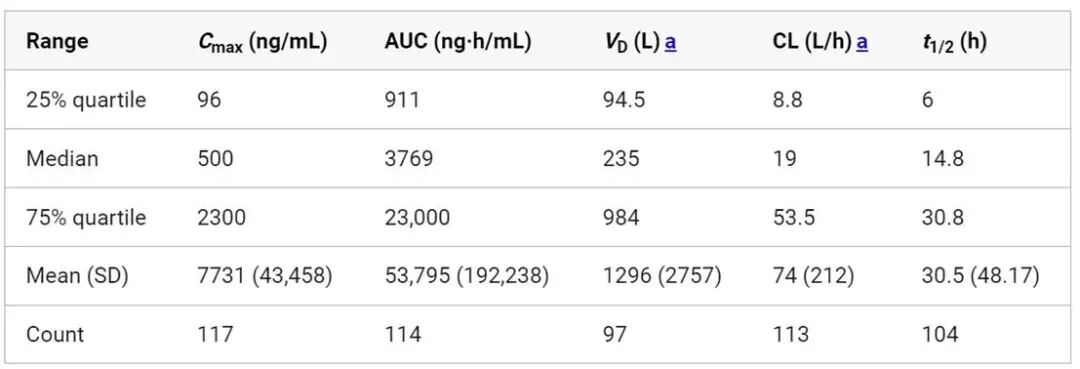
对药物代谢动力学参数的深入分析进一步揭示了现代口服药物的多样性特征。这些药物的中位血药浓度峰值(Cmax)为500 ng/mL,中位药时曲线下面积(AUC)为3769 ng·h/mL,中位分布容积为235 L,中位清除率为19 L/h,中位半衰期为14.8小时。这些参数为早期药物研发中的目标设定提供了有价值的参考基准。
表. 获批药物人体的药代动力学性质
特别引人注目的是血浆蛋白结合率的数据:高达58%的药物血浆蛋白结合率超过95%,其中29%更超过99%。尽管高蛋白结合在传统观点中可能被视为限制因素,但现实中并未阻碍这些药物的成功上市。
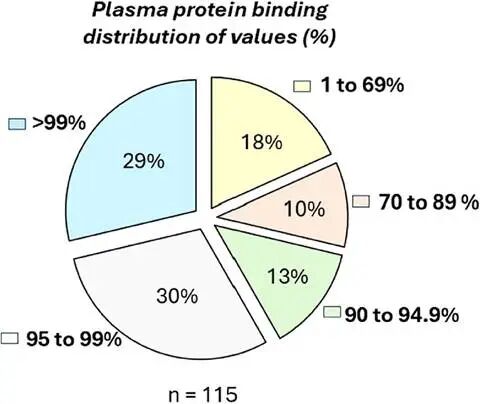
获批药物, 组合物及活性代谢物的人血浆蛋白结合率统计
更有若干药物展现出“非典型”药代行为,如沙诺美林(Xanomeline)的口服生物利用度仅为1%,清除率高达1950 L/h;奥扎莫德(Ozanimod)和加奈索酮(Ganaxolone)的清除率也分别达到192 L/h与430 L/h。
另一个值得关注的例子是索格列净(Sotagliflozin),其清除率为300 L/h。这些案例共同提示,即使某些药代参数偏离传统理想范围,只要在目标疾病中展现出足够疗效与可控的安全性,药物仍可能获得监管机构的批准。
安全警示:黑框警告与禁忌症不容忽视
在安全性方面,分析显示有22%的药物附带有黑框警告,42%列有禁忌症,16%存在QT间期延长相关警示。若按治疗领域细分,神经科学类药物中带有黑框警告的比例为28%,心血管药物、自身免疫药物和肾脏疾病药物中也占有较高比例,而肿瘤药物中这一比例为14%。
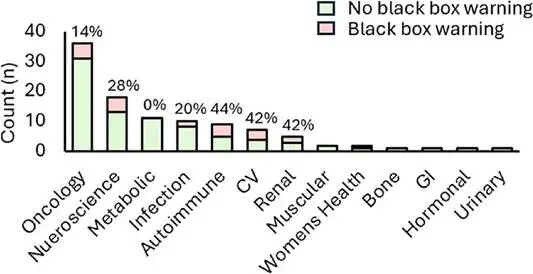
带有黑框警告的药物在各治疗领域中的占比
常见的黑框警告内容涵盖心血管风险、胚胎-胎儿毒性、自杀倾向及严重感染等。这一现象并不令人意外,因为这些领域往往作用机制新颖、靶点复杂,风险-获益比的评估标准也不同于常见慢性病药物。
与此同时,药物-药物相互作用(DDI)也是临床使用中需高度关注的问题。其中,CYP3A4是最常涉及的代谢酶,62种药物提示与其诱导剂存在相互作用,48种提示与抑制剂合用需谨慎,30种药物自身是CYP3A4的底物。
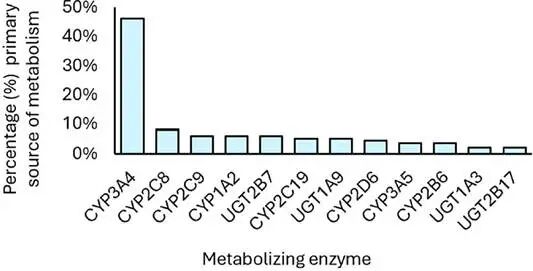
获批药物的主要代谢酶
此外,P-gp和BCRP等转运蛋白相关的DDI也较为常见,分别涉及22种和12种药物。尽管广泛的DDI可能限制部分药物的使用场景,但在缺乏替代疗法的疾病中,此类风险仍可通过临床监测与管理得以控制。
活性代谢物:隐藏在体内的“第二战场”
活性代谢物的普遍存在是另一个值得深入探讨的议题。在所分析的104种药物中,至少有14种会在体内转化为具有药理活性的代谢产物。
其中,N-脱烷基化是常见的代谢路径,如司美替尼(Selumetinib)通过CYP3A4代谢生成N-去甲基司美替尼,尽管其在血浆中的水平不足母药的10%,但其药理活性更强,贡献了21-35%的总药理活性;
瑞普替尼(Ripretinib)在CYP3A4、CYP2C8和CYP2D6作用下失去N-甲基生成DP-5439,其活性与母药相当;莫博赛替尼(Mobocertinib)和英菲格拉替尼(Infigratinib)也通过类似途径生成多个活性代谢物。
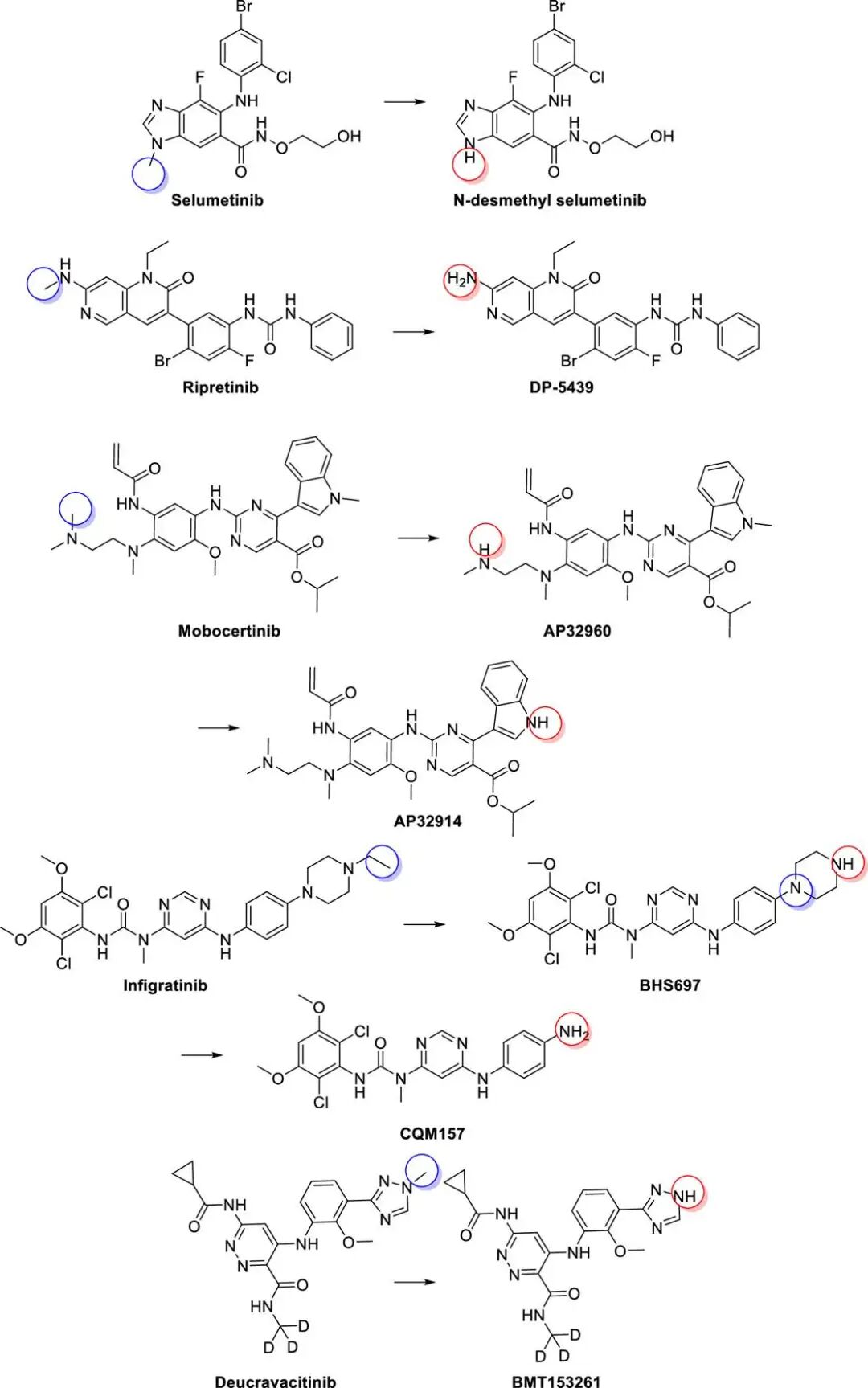
N脱烷基生成活性产物的药物实例
非N-脱烷基化途径同样值得关注。菲啶硝唑(Fexinidazole)通过硫原子的单氧化和双氧化生成M1和M2两个活性代谢物,其AUC值分别是母药的11倍和34倍;
阿伐可泮(Avacopan)通过对位芳香甲基的CYP介导羟基化生成活性代谢物;阿布昔替尼(Abrocitinib)在丙基侧链上发生氧化生成两个活性代谢物。
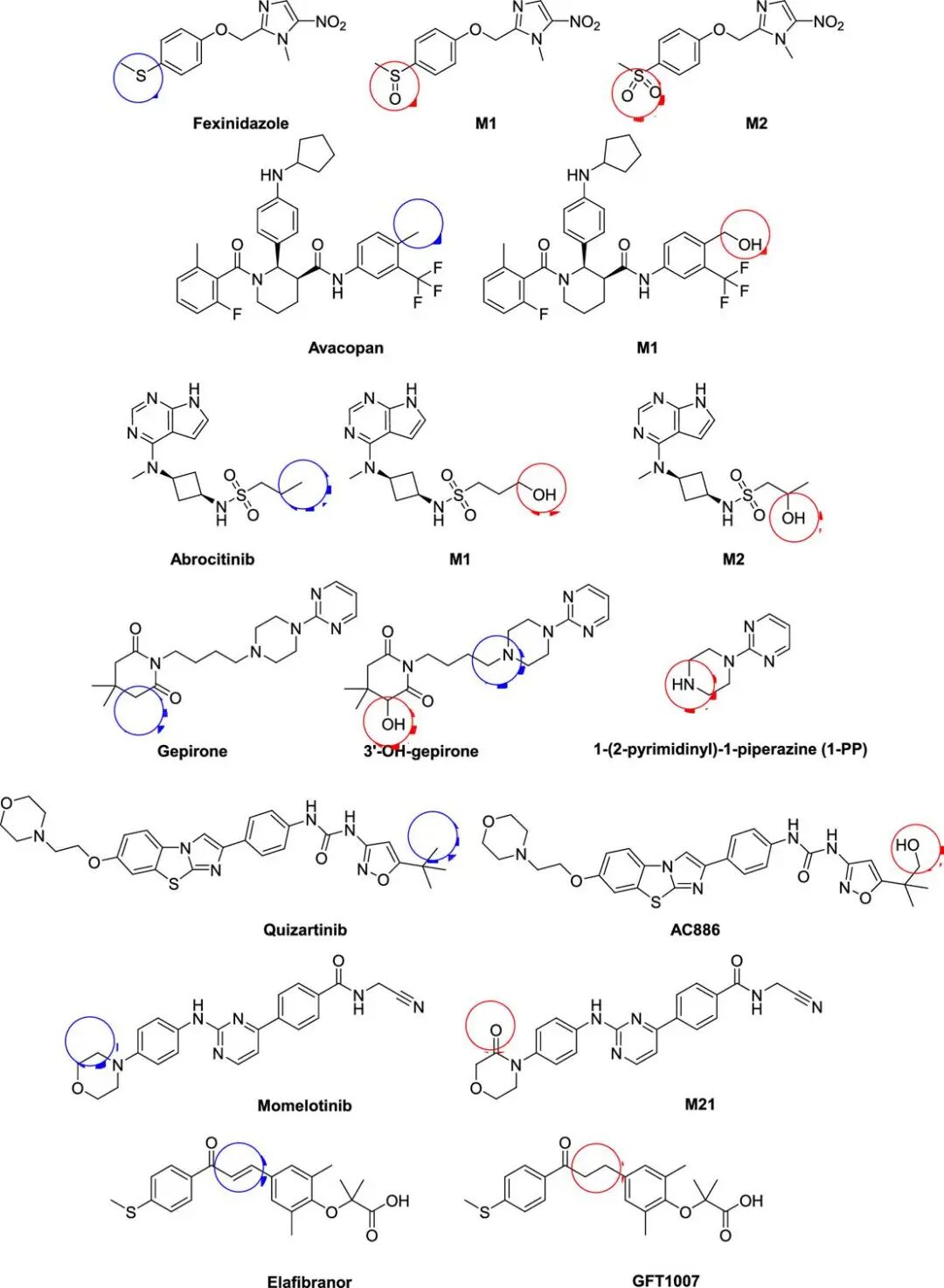
原型药物及它们的活性代谢产物
非典型化学结构:挑战传统认知
从药物化学的角度来看,本综述中出现的多种“非典型”化学结构也挑战了传统药物化学中的某些教条认知。
例如,来那卡帕韦(Lenacapavir)分子中的炔烃结构,帕罗瓦罗汀(Palovarotene)中的苯乙烯片段,以及若干药物中出现的N–O键(如吉维诺司他(Givinostat)中的羟肟酸)、芳香溴、硝基(尼夫替尼(Nifurtimox)、奥匹卡朋(Opicapone))及N-氧化物(菲啶硝唑、阿里莫洛)等,在过去可能被视为具有代谢不稳定性或化学反应性风险的结构。
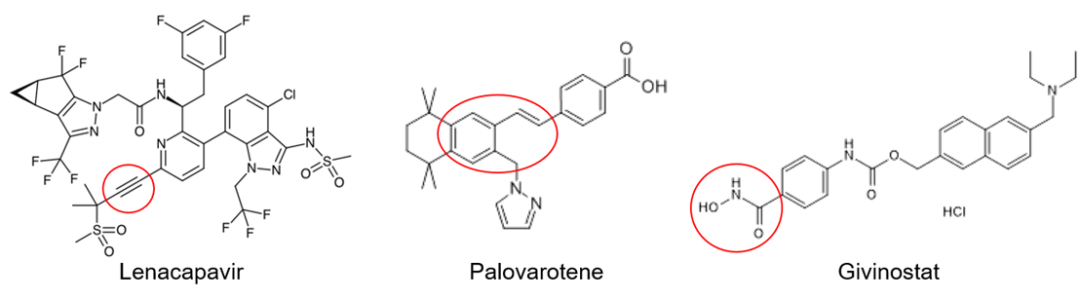
获批分子中存在的传统分子设计中不受欢迎的结构片段
然而,它们在实际应用中仍成功获批,说明任何官能团的安全性都与其所处的整体化学环境密切相关,不应被孤立判断。特别值得注意的是,某些官能团如硝基在尼夫替尼中本身就是其抗寄生虫作用机制的核心部分。
设计原则:打破教条,拥抱多样性
综合以上分析,作者提出两项核心建议:
- 其一,应避免制定过于僵化的候选药物标准,以免将那些具有一定非典型特性、却可能为患者带来重大收益的化合物过早淘汰。现实中,高剂量(39%的药物日剂量超过300毫克)、高蛋白结合、低口服生物利用度、复杂代谢特征乃至一定的安全性风险,都未阻止这些药物最终成功上市。
- 其二,药物化学家应掌握基本的人体药代预测与剂量估算方法,包括异速缩放(如从大鼠到人的清除率推算)、体外-体内外推(如基于肝微粒体数据预测体内清除率)、目标受体覆盖分析(如IC50、IC75、IC90对应的游离药物浓度)等,从而能够在项目早期对化合物的开发潜力做出科学评估,而非依赖于“这永远成不了药”的直觉判断。
如今,一些公开可用的计算工具如Molmatinf的“药物化学工具箱”等PK预测工具,也为早期评估提供了便利。
结论:多样性是创新的土壤
总而言之,这篇综述通过对近年获批口服药物的全景式分析,深刻揭示出成功药物的特征具有显著的多样性,无法用单一标准简单界定。在104种药物中,42种为首创药物,59种为孤儿药,凸显了小分子药物在解决未满足临床需求中的持续创新潜力。
对药物研发者而言,这份分析既是一面镜子,反射出现实中药物的真实面貌;也是一把钥匙,鼓励我们在坚持科学原则的同时,保持结构的开放性与思维的批判性,避免被过于僵化的教条所束缚。
正如作者所言,在未来的药物发现之旅中,我们或许应当少问“这能成为药吗?”,而多思考“为什么不能?”,以此推动更多真正改变患者生命的创新疗法走向临床。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-11-22,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号