单细胞测序不同亚群的特异性基因的计算新选择 starTracer

单细胞测序不同亚群的特异性基因的计算新选择 starTracer

生信技能树
发布于 2024-11-21 10:02:48
发布于 2024-11-21 10:02:48
众所周知,单细胞转录组有两种差异分析,首先是不同单细胞亚群之间的,比如t细胞和b细胞,其次是同一个单细胞亚群在不同生物学条件下面的差异比如癌症和癌旁组织的t细胞的差异,这两个差异分析有的方法学区别很大!
- 不同单细胞亚群之间的差异分析:
- 目的:比较不同细胞类型(如T细胞和B细胞)的基因表达模式,以揭示它们独特的生物学特性和功能。
- 数据特点:通常涉及多个细胞亚群的比较,每个亚群可能包含数百到数千个单细胞。
- 同一单细胞亚群在不同生物学条件下的差异分析:
- 目的:探究同一细胞亚群(如T细胞)在不同生物学条件(如癌症和癌旁组织)下的基因表达变化,以理解环境或疾病状态对细胞功能的影响。
- 数据特点:通常涉及同一细胞类型的内部比较,比较的是同一细胞亚群在不同条件下的表达差异。
区别:
- 生物学背景:第一种分析更关注细胞类型的固有特性,而第二种分析更关注环境或疾病状态对细胞功能的影响。
- 统计模型:虽然两种分析都使用统计测试,但第二种分析可能需要更复杂的模型来控制混杂因素,如疾病进展、治疗历史等。
- 生物学解释:第一种分析的生物学解释侧重于细胞类型的分类和特征,第二种分析则侧重于特定条件下的生物学变化和潜在的分子机制。
- 应用:第一种分析有助于理解细胞异质性和发育轨迹,第二种分析有助于揭示疾病过程中的分子变化和治疗靶点。
在实际操作中,两种分析方法可以根据研究目的和数据特点进行选择和调整,以获得最有意义的生物学信息。
通常情况下,不同单细胞亚群之间的差异(如T细胞和B细胞)往往比同一单细胞亚群在不同生物学条件下的差异(如癌症和癌旁组织的T细胞)更容易区分。
因为不同单细胞亚群,比如T细胞和B细胞是两种功能和发育路径截然不同的免疫细胞,它们具有不同的基因表达谱和生物学功能,因此它们之间的差异通常非常明显。
但是如果生物学重复的数量不足,那么随机变异可能会掩盖同一个细胞亚群在不同生物学条件下面的真实的生物学差异。同一细胞亚群在不同条件下的差异可能需要更精细的分析方法,包括复杂的统计模型和数据预处理步骤,以区分真实的生物学信号和噪声。同一细胞类型在不同条件下可能涉及复杂的生物学过程和调控机制,这些过程可能在基因表达水平上不易被察觉。
关于前者,我们推荐过:神兵利器——单细胞细胞类群基因marker鉴定新方法:COSG,因为Seurat 官方的 FindMarkers与FindAllMarkers函数,非常的耗时如果细胞数量比较多,除非搞多个线程去并行运算,但是如果并行就会超级耗费内存!所以我很早就放弃了它,现在给大家的代码里面都是COSG包啦。但是最近武汉大学的小伙伴们“毛遂自荐”了她的工作:
接下来我们介绍一下:starTracer:一个高效、准确的单细胞测序标志基因检索软件/R包
key words: starTracer, single-cell RNA-seq; marker gene; R package.
关键词: starTracer;单细胞测序;标志基因;R包.
原文链接:starTracer is an accelerated approach for precise marker gene identification in single-cell RNA-Seq analysis[1]
1 背景介绍
有幸拜托Jimmy老师和生信技能树团队,借此平台,为我们的工作做一个介绍,让更多科研工作者的单细胞分析变得灵活而高效,也希望能够为我们的工作指出bug,精益求精。
这是关于R包starTracer的设计过程的思路、部分结果的呈现,文末附有使用方法的中文指导。具体的方法部分和完成的结果,还请大家移步原文。我将会主要在这里分享在论文中没有被记录的部分。如果在使用过程中有发现任何问题,请到我们的github page进行反馈,我希望帮助每一个愿意使用我们软件的科研工作者,在你们的数据上得到满意的结果。十分感谢!
2 问题引入
不知道大家在进行单细胞测序的标志基因的计算的时候,是否面临了以下几个问题:
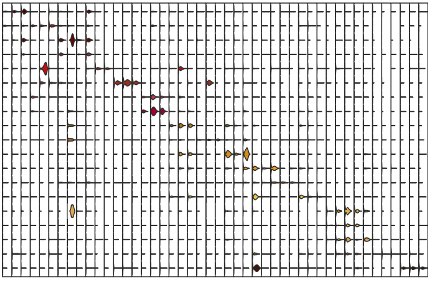
a. 计算时间耗时长,有时候10万个细胞的样本需要花费40分钟来计算得到结果。(用40分钟来摸鱼休息其实也是不错的) b. 计算得到的标志基因的效果不佳。尤其是当我们对一个Major clust进行进一步亚群的划分时出现。这个时候Marker基因常常会呈现出一个“糊了满屏”的感觉:即特异性程度很低。这里给大家展示一下当时我在处理自己的数据的时候遇到的问题:

图,来自兴奋性神经元的sub-cluster的标志基因的小提琴图。使用Seurat包的FindAllMarkers函数获取,按照p-value进行筛选后,使用logFC进行排序。由于涉及未发表数据,仅选取部分作为展示使用。
3 解决方案:补丁
计算时间过长速度过慢,我想这是R的硬伤。所以,基于C++的Presto[2] 便是一个不错的解决方案,这是一个用于加速Marker基因寻找的不错的工具,但是Seurat V4并没有纳入它,不过目前在网络上可以下载的Presto已经支持直接读取Seurta对象,对V4用户友好。而V5版本则是直接纳入了Presto。我们需要注意的是,Presto仅仅是对wilcox等统计学分析过程进行加速,其本身并不会改变FindAllMarkers的计算结果。
而计算的效果不佳,我想这是很多研究人员在针对细分亚群的标志基因的时候面临的问题。常见的解答方案有以下几种(为了方便,我直接插入图片,大家感兴趣的可以去查阅原文):补丁1,计算一个指标,用于衡量其特异性(来自Allen Brain Institute)
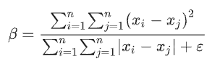
Hodge et. al., 2019, p. 16. For gene i, the overall specificity level is measured by:sum(distance^2)/ sum(distance)0 ~ 1. low specificity; high specificity
补丁2,计算一个指标,同样用于衡量特异性(实际上这个算法是在Stimulated Single Cell Data里面去生成Ground Truth Marker)
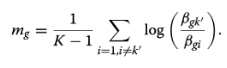
Pullin ,McCarthy, 2024, p. 28. For gene g, the specificity level in cluster k’ is measured by:sum of the log-fold changes of gene g in cluster k’ comparing to all the rest of the clusters divided by total number of clusters (K) .
补丁3,我的方法:(其实灵感来源于Seurat FindAllMarkers 函数的pct1和pct2)
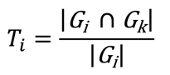
Feiyang et. al., 2024. For gene i, the overall specificity level in cluster k is measured by:For gene i, the intersected number of clusters that with gene i as marker and that expressed gene i, divided by the number of clusters that expressed i gene.
当然,Seurat本身自己也提供了pct.1、pct.2以及logFC,Jimmy的生信技能树团队的推文对此有详细的介绍。限制pct以及限制FoldChange,本身都是一个不错的办法,但是Seurat的FindAllMarkers函数的过长的计算时间(10w个细胞,在我们很不错的芯片上,仍然需要计算40分钟;60w个细胞则需要花费6个小时)让这几个参数陷入一个使用上的困境:研究人员在得到结果之前,是不知道什么样的logFC和pct.1、pct.2是最好的,而只能够选择一个相对宽松的限制——使用过高的log2FC限制了特异性后,可能会导致自己忽略掉一些可能表达水平更高的基因(可以参考原文里关于基因表达水平和特异性的论述)。所以往往大家还是采用宽松的限制,花更多的时间来得到一个巨大的表格,再进行自己的手动筛选——例如上述的各种补丁。
而我在“发明”了补丁3后,标志基因的效果得到了大幅度的提升,当然,总体表达水平出现了降低(图中没有体现):
图,过滤后的标志基因,我们发现基因的总体表达水平得到了降低,但是实际上相对的表达量获得了提高。同样的,为了数据隐私性,我把行和列名字给隐去了。
“基因的总体表达水平得到了降低,但是实际上相对的表达量获得了提高”这个理念,我认为是来自于生物学本身的固有本质——这让研究人员想要找到更有特异性的表达结果时来筛选精细的细胞亚群的时候,则必然从更低表达量的基因的入手,而这进一步要求更高的实验技术水平的支持。这一逻辑本身也是合理的。
4 更加“优雅”的解决方案
虽然计算速度、特异性的问题仿佛得到了解决,但是总是通过打补丁的方式:例如用Presto使用C++来对计算进行加速,这本身并不优雅,而是依赖于计算机的暴力、利用三个补充方程,不仅没有简化流程,反而让计算过程变得更加复杂。其实曾经在初学单细胞测序的时候,Jimmy老师的博客里面有一篇文章对于FindAllMarkers函数的log2FC的解释让我记忆犹新。于是我去翻阅了FindAllMarkers的源码 (当时是我第一次尝试去解读R包的源代码,感谢知乎大佬们的铺垫),对此进行了佐证。我发现其实不论是Seurat,还是Scanpy、Monocle 3,大家都在使用的一个思想是:
对任意的基因,在计算的过程中,软件将会将其放进某一个细胞类型,再与其他的所有的细胞类型的总和的进行差异比较。
而我不赞同使用这个思想来寻找标志基因,因为使用这个方法得到的结果,在条件比较“艰难”的时候,效果很差。具体的弊端体现在如下几个方面:
4.1 现有方案的弊端1: Dilution Issue
FindAllMarkers的计算逻辑是循环调用FindMarkers,在找marker的时候,我们用了FindAllMarkers;在后续找差异基因,我们用的是FindMarkers。在这里,大家不知是否已经产生了疑问:这里我们是否搞混淆了一个概念/或者是遗漏去辨析了两个概念:marker基因和DEG是不一样的:
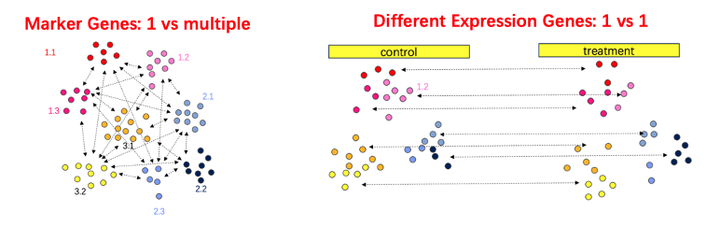
图,左:理想的marker基因的寻找鉴定逻辑:一个细胞亚群的标志基因,应该由其与剩余的细胞亚群逐个对比获得参数,并综合考虑参数而确定。右:DEG,由相同的细胞亚群跨越condition互相比较而确定。
在理清了这个观点后,我们不难发现,上述的三个“补丁”方程,其实都是把某一个亚群逐个地与剩余所有亚群比较,并考虑总体结果来进一步地衡量基因在一个亚群的特异性程度。有意思的是,如果我们去查看上述的“补丁”1、2的原文,我们会发现,作者们的字里行间都透露了一个信息:Binary Expression. (不清楚翻译是否正确,含义大概是:二分类似的表达——即,在一个亚群高表达,而在别的亚群都是低表达)。这不是一个废话,因为FindAllMarkers的计算逻辑里面,其他的所有的亚群都被平均了,这将会导致一个很严重的“稀释”问题——即原文里所描述的dilution issue(感谢QBI赵老师帮助总结了这个清晰易懂的概念)。
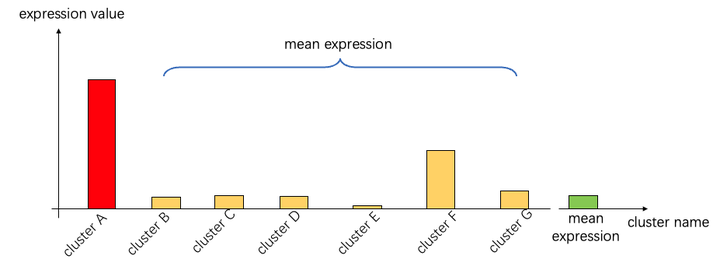
图:Dilution Issue图解,即一个细胞亚群(clusterF)在平均化过程中,表达水平被其他表达更低的亚群拉低了。导致log2FC呈现出很大的值,而实际上该基因由于其在F当中存在中等程度的表达,并不能够作为ClusterA的标志基因/或者是在排序上需要往后稍稍,把top的位置留给更好、特异性更高的基因。
4.2 现有方案的弊端2: 冗余计算
不知道大家是否在FindAllMarkers的时候遇到过一个问题:假设20个细胞亚群,在按照pvalue过滤和log2FC排序后,如果直接按照cluster分组,再分别选取前20个基因的方案(df %>% group_by(cluster) %>% slice_head(n = 20))来找每个亚群的top10 marker,为什么得到的dataframe的数量有时候却少于20 * 10 = 200 个?
这是因为FindAllMarkers有时候将一个基因鉴定成为了多个cluster的marker基因(并且他们还都是显著的、log2FC很高的)——为了解决这个问题,我们需要首先去选择log2FC最高的那一个,把其作为这个marker的真实的cluster。
这种“打补丁”一样的去除多余信息的操作,暗示这实际上是源自于FindAllMarkers产生了大量冗余的计算:对于一个基因A,FindAllMarkers将会分别把它放到1~N每一个cluster里面,再和其他的细胞亚群的平均进行计算。虽然实际的运算流程并非一个个的基因去计算,但是针对每一个基因,仍然产生了多余的N-1次运算——因为任何一个基因A(在我们不考虑其客观事实上是否是标志基因的情况下)它只有可能在其表达水平最高的那个细胞类型中成为标志基因(这个想法和上面的“选择log2FC最高的那一个”其实是一致的)。既然如此,那我们为什么不在进行复杂的参数运算之前,就告诉软件:“这个你不用算了,基因A不可能是细胞B的marker基因,因为基因A的最高表达在细胞C当中,除此之外的其他细胞亚群,你都不需要计算。”
4.3 对于补丁3的完善:限制表达水平
补丁3的结果大家可以看到,marker基因的表达水平实在是有点低。为什么太低的表达量不好?这有诸多方面的原因:
- 由于10x genomics的测序方案限制(每个beads里面捕获到的RNA fragment的数量远小于bulk RNA-seq),在细胞数量太少的时候,基因的表达水平会出现偏差。为了解决特异性程度太高导致的表达水平降低的问题,我们需要在寻找标志基因的时候提供一个参数,来限制表达水平,以便于有时候让我们能够牺牲特意度来获取更高的表达量。当然如果你是使用的smart-seq这种测序,我认为受到的影响可能会相比10x genomics的影响更小。
- 10x genomics的测序,不论是单细胞核测序还是单细胞质测序,低表达量的基因,有可能是系统误差产生的(个人猜想,未经验证)。
4.4 思路小结
综上所述:我们的设计思路便是
- 对基因表达进行二分类定义
- 提前将基因分配到其唯一“有可能”成为marker基因的cluster当中
- 在计算时,允许研究人员以牺牲特异度的方式来获取表达量
对于设计的总体逻辑,我们在Supplementary Figure 1 中作出了详细的图解,整个计算逻辑的核心都是基于初等代数。对于设计的细节部分,大家可以参考Methods部分,在此我们不再展开。
5 结果呈现
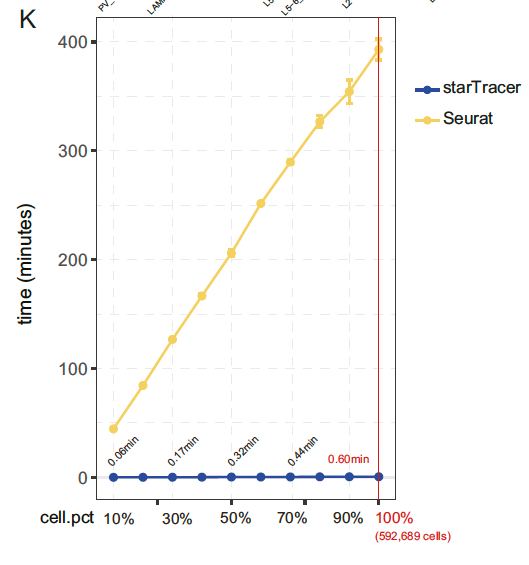
详细的结果,仍然请大家移步原文。在这里我们仅对结果作部分的解释:这里我们将一个具有60万细胞的数据进行了拆分,按照10%~100%的数据量,分别计算了默认参数下的Seurat的FindAllMarkers的计算时间和starTracer的searchMarker(searchMarker将会是你在使用starTracer这个包的时候,具体使用的函数,具体信息请看文末)的计算时间。我们可以看到starTracer的计算时间随样本量的扩大而增加的斜率是明显小于Seurat的。在60万个细胞的时候,在我们的服务器上,Seurat甚至需要花费6小时来得到结果,而starTracer只需要30余秒。我们将整个计算速度提高了数百倍。
我们也与C++选手Presto进行了比赛,在使用Seurta原装Average Expression函数的时候,我们的计算速度实际上没有超越Presto。后来,参考了Zachary DeBruine教授在社区慷慨贡献自己的Average Expression函数 ,让我们的计算速度超越了Presto。对此深表感谢。如果还有高手可以将starTracer装上C++发动机,我想我们会跑的更快。
当然,starTracer的计算的结果也是相当的不错:
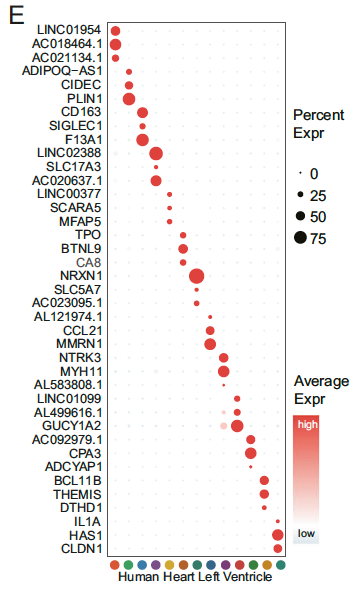
从计算结果上来看,提升最明显的是针对神经元当中的抑制性神经元的标志基因。我们纳入了所有的基因进行标志基因的寻找,发现starTracer在抑制性中间神经元的提升相当明确(当然,我们已经知道这些神经元的标志基因了),例如SST作为SST中间神经元的标志基因,PVALB作为PVALB中间神经元的标志基因,VIP作为VIP中间神经元的标志基因,NDNF作为NDNF中间神经元的标志基因。
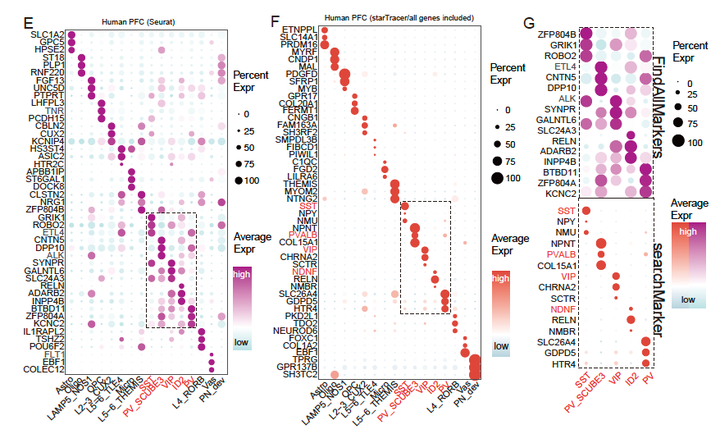
当然,我们随后也是用Simulated Data来对FPR进行了系统性的评估。评估了starTracer在低数量细胞上的稳定性以及复杂注释下的表现能力。其他的内容,请大家移步原文。
6 使用方法
具体的使用方案请参考github主页:starTracer[3] 下载:
if (!requireNamespace("devtools", quietly = TRUE)){
install.packages("devtools")}
devtools::install_github("JerryZhang-1222/starTracer")
在这里也作一个简单的介绍:你可以输入一个Seurat对象、一个dgCMatrix或者是一个Average Expression Matrix。返回的结果是一个list(目前没有使用S4类,只是简单地用了一个list)。
list里面包含:
para_frame:包含所有中间参数的data frame,其中max.X列说明了该marker在何种细胞中被认为是marker基因。n是衡量程度的一个指标,为1的是最佳。在相同的n中,MI大,其作为marker基因的能力越好。rest_clust说明了当n不等于1时,其他有可能对marker的特异性构成影响的其他细胞类型。
genes.markers:marker基因
expr:Average Expression Matrix
res <- searchMarker(
x = x, # 输入一个Seurat对象、一个dgCMatrix或者是一个Average Expression Matrix
thresh.1 = 0.5, # 建议使用默认值
thresh.2 = 0.3, # thresh2越高,表达水平越高,特异性越低。请设置在0-1范围内
method = "pos", # 我们提供了寻找negative的方案,当然这目前只是一个alpha版本,如果需要,请设置为"all"
num = 2, # 输出的top N marker基因数量
gene.use = NULL, #留空使用所有基因,设置为“HVG”则使用HVG
meta.data = NULL,
ident.use = NULL
)
#您可以使用以下代码来获取marker基因的pseudo-bulk表达矩阵
res$expr[res$genes.markers,]
#您可以使用以下代码来获取marker基因记忆其对应的cluster
res$para_frame[res$genes.markers,c("max.X","gene")]
其他下一步可以改进的方向
输出部分比较简陋,代码没有封装非常完善
针对一个cluster内部的基因表达抑制性没有作过多的阐述
缺乏python等其他平台版本
可能会有不少bug
一些想法
再次向生信技能树团队、Jimmy老师表达谢意。不论是这个idea的产生还是实现,你们的无私的奉献都给予了我很大的帮助。这个软件实际上所有的内容几乎都是初等代数的东西,也很有可能有更多的改进的空间。由于作者本身不是数学和计算机专业出身,所以在代码的规范性上或许还有很多值得改进的地方,希望大家能够给予意见。
另外如果有帮助到读者您,请考虑引用我们的工具。谢谢!
Zhang, F., Huang, K., Chen, R. et al. starTracer is an accelerated approach for precise marker gene identification in single-cell RNA-Seq analysis. Commun Biol 7, 1128 (2024). https://doi.org/10.1038/s42003-024-06790-6
参考资料
[1]
原文链接:starTracer is an accelerated approach for precise marker gene identification in single-cell RNA-Seq analysis: https://www.nature.com/articles/s42003-024-06790-6
[2]
Presto: https://doi.org/10.1101/653253
[3]
starTracer: https://github.com/JerryZhang-1222/starTracer
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-10-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号