Cell. Syst. | 一种端到端的自动化机器学习工具,用于解释和设计生物序列

Cell. Syst. | 一种端到端的自动化机器学习工具,用于解释和设计生物序列

DrugOne
发布于 2023-09-19 14:58:57
发布于 2023-09-19 14:58:57
编译 | 曾全晨 审稿 | 王建民
今天为大家介绍的是来自James J. Collins团队的一篇论文。自动化机器学习(AutoML)算法可以解决将ML应用于生命科学时面临的许多挑战。然而,由于这些算法通常不明确处理生物序列(如核苷酸、氨基酸或糖肽序列),且不容易与其他AutoML算法进行比较,它们在系统和合成生物学研究中很少被使用。在这里,作者介绍了BioAutoMATED,这是一个用于生物序列分析的AutoML平台,将多个AutoML方法集成到一个统一的框架中。用户可以自动获得分析、解释和设计生物序列的相关技术。BioAutoMATED可以预测基因调控、肽-药物相互作用和糖肽注释,并设计优化的合成生物学组件,揭示突出的序列特征。通过自动化序列建模,BioAutoMATED使生命科学家更容易将ML应用到他们的工作中。

近年来大规模高维生物学数据集的出现促进了机器学习(ML)方法在研究和预测生物现象方面的广泛应用,为基因组学带来了令人振奋的突破,也在系统生物学、合成生物学和结构生物学等领域带来更多的前景。中等到大规模的生物学序列数据集,包括核酸、肽和糖链序列等,无处不在。在这些数据集上使用机器学习可以帮助研究人员提取生物学见解,并加快设计具有所需性质的序列。
计算分析和机器学习技术通过在线教程、开源代码、交互式笔记本和软件包变得更加易于科学家使用。然而,构建、训练和部署ML模型通常需要ML专业知识。各种用户决策可能会极大地影响ML模型的质量和性能。了解哪些设计决策重要,以及如何为任何给定的数据集做出最合适的决策,对于在ML经验有限的生命科学研究人员尤为重要。即使在熟练的ML实践者中,选择算法技术和调整模型参数(通常从几千个到数亿个不等)也是困难的。事实上,手动定义模型和模型参数优化需要相当的专业知识和时间来实现,并且在生物学和生物医学的许多应用ML研究中可能只提供有限的好处。
使ML更容易应用于分析生物数据集的一个有前途的途径是使用自动化机器学习(AutoML)。AutoML包含自动化设计和部署ML流程的方法,减少用户干预。端到端的AutoML将为生命科学家提供简单的数据预处理、特征提取、模型选择和优化以及性能评估。AutoML技术可以自动识别适当的模型架构(模型中使用的算法类型)和模型超参数(可以调整的参数,影响模型性能)。由于确定最佳的模型架构和超参数值对于ML专家来说通常也是困难的,适当实现的AutoML策略可以帮助生命科学家构建初始的预测模型。此外,AutoML也可以为更有经验的ML实践者提供一种快速生成基准模型以进行比较或快速识别具有令人鼓舞性能的广泛模型类别的方法。
目前有许多丰富多样的AutoML工具可用。许多成熟的AutoML工具仅搜索神经网络模型类别。然而,最令人兴奋的AutoML工具之一是基于树的优化方法,它们搜索“浅层”或更简单的scikit-learn模型,如随机森林分类器。这些技术对于比神经网络更小、更稀疏的生物学数据集可能更合适,但它们尚未与神经网络结构搜索方法共同用于加速生物学序列分析。事实上,模型的架构选择对于模型性能很重要,最近的研究表明没有单一“最佳”的AutoML工具,强调在一个平台上评估许多类别的模型的重要性。
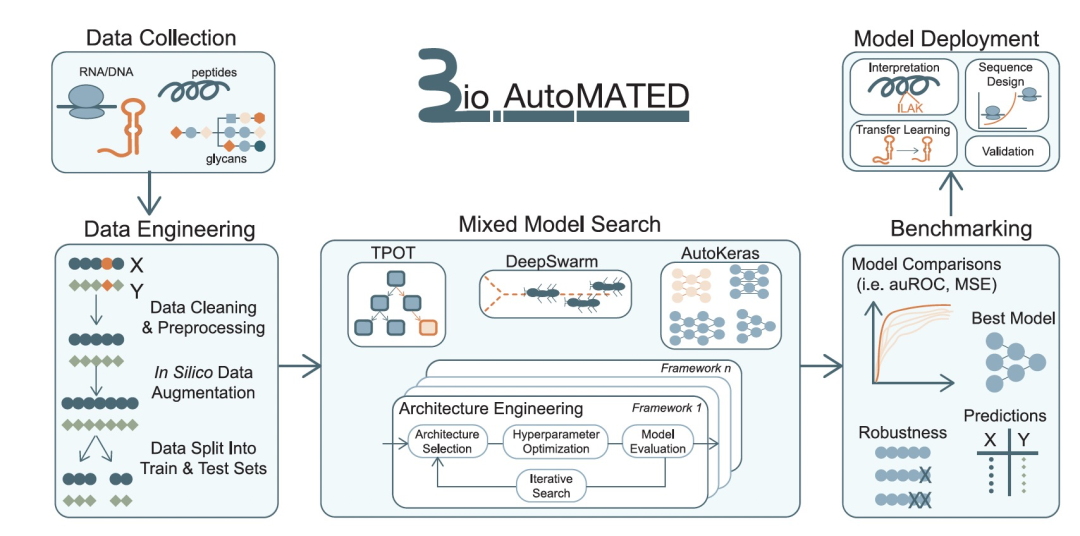
图 1
因此,作者在这里展示了生物自动化机器学习工具用于解释和设计,即BioAutoMATED,这是一个优化用于构建核酸、肽和糖链序列输入模型的端到端AutoML框架(图1)。BioAutoMATED系统整合了多个开源AutoML工具,同时还采用了各种不同的搜索机制,允许比之前报道的更广泛的架构搜索空间。作者将此集成与自动数据导入、预处理、架构选择、超参数搜索、模型训练、模型部署和性能报告相结合,全部嵌入在一个易于使用的高级编程接口中,可以在Jupyter Notebook中访问。作者展示了BioAutoMATED通过自动使用预测序列中显著区域和模体的技术来促进生物模型解释。
BioAutoMATED自动化模型过程
用户的数据集可以包含任意数量的给定类型的生物序列(例如核酸、氨基酸或糖链)。为了获得最佳结果和合理的时间范围,作者建议数据集包含1000到50万个序列,并且序列长度不超过几百个“亚基”(核苷酸、氨基酸或糖单糖及连接键)。用户上传所选数据集后,BioAutoMATED将在相对无需干预的情况下继续进行。在选择一小组用户定义的选项后,例如序列类型、允许每个AutoML搜索运行的时间以及所需的预测任务(回归、二分类或多类分类),BioAutoMATED将进行所有后续的数据预处理,包括清理格式不正确的输入。输入序列的字母表,或者是出现在用户序列数据集中的核苷酸、氨基酸或单糖和键的集合,将被自动推断。例如,肽序列的字母表由出现在输入数据中的所有氨基酸组成,这允许学习输入字母的相对重要性,而不是假设数据集中包含所有氨基酸。然后,这个字母表用于为所有输入序列生成向量表示。
经过自动数据预处理后,BioAutoMATED通过实施DeepSwarm、AutoKeras和Tree-based Pipeline Optimization Tool (TPOT)的修改版本来执行AutoML模型搜索,以共同搜索不同的架构和超参数。BioAutoMATED生成这三个AutoML系统的结果,可以进行联合评估和比较。一个关键原因是我们的集成方法仅依赖于标准化的输入和输出。因此,通过调整当前平台,其他AutoML系统也可以被合理地整合进来。
BioAutoMATED适用于不同的数据类型、大小和处理选项
BioAutoMATED自动提供了多种选项,让用户可以更多地参与模型选择、控制和数据集扩展。例如,系统生物学研究者可能不确定训练典型ML模型所需的数据量,因为这取决于多种因素,例如预测任务的难度、数据的质量和序列空间的维度。为了评估训练数据的量是否足够优化模型,用户可以选择运行数据消融实验,使用训练在随机选择的数据集上的模型,并逐渐减少数据集的大小。此外,BioAutoMATED平台还自动计算每个AutoML阶段(如架构搜索、数据消融研究、乱序对照等)所花费的时间,用户可以对其进行评估。对于核酸序列,用户还可以选择通过计算互补序列、反向互补序列或两者来增强数据集,并将这些合成序列与原始序列的标签一起加入到数据集中。这个增强步骤受到图像分类中数据增强的启发,在图像分类中,可以通过裁剪、旋转或转换原始图像来人工增加数据集的大小。在一些情况下,对于小型训练数据集,这个数据增强步骤可以提高模型性能,特别是对于那些互补序列或反向互补序列被期望具有与原始序列类似含义的序列而言。
由于训练集中的所有序列必须具有相同的长度,以便与三种AutoML工具兼容,BioAutoMATED提供了几种选项来标准化具有不同序列长度的数据集。序列可以填充到最大长度、截断到最小长度或标准化为平均序列长度。为了扩展BioAutoMATED模型的适用性,用户提供了一个迁移学习模块,新数据可以用于“微调”现有模型。用户可以使用额外的数据重新训练BioAutoMATED生成的任何现有DeepSwarm、AutoKeras或TPOT模型,进一步增强模型的预测能力,即使数据集在大小和质量上不断增长。
BioAutoMATED阐明了用于基因调控的核糖体结合位点设计
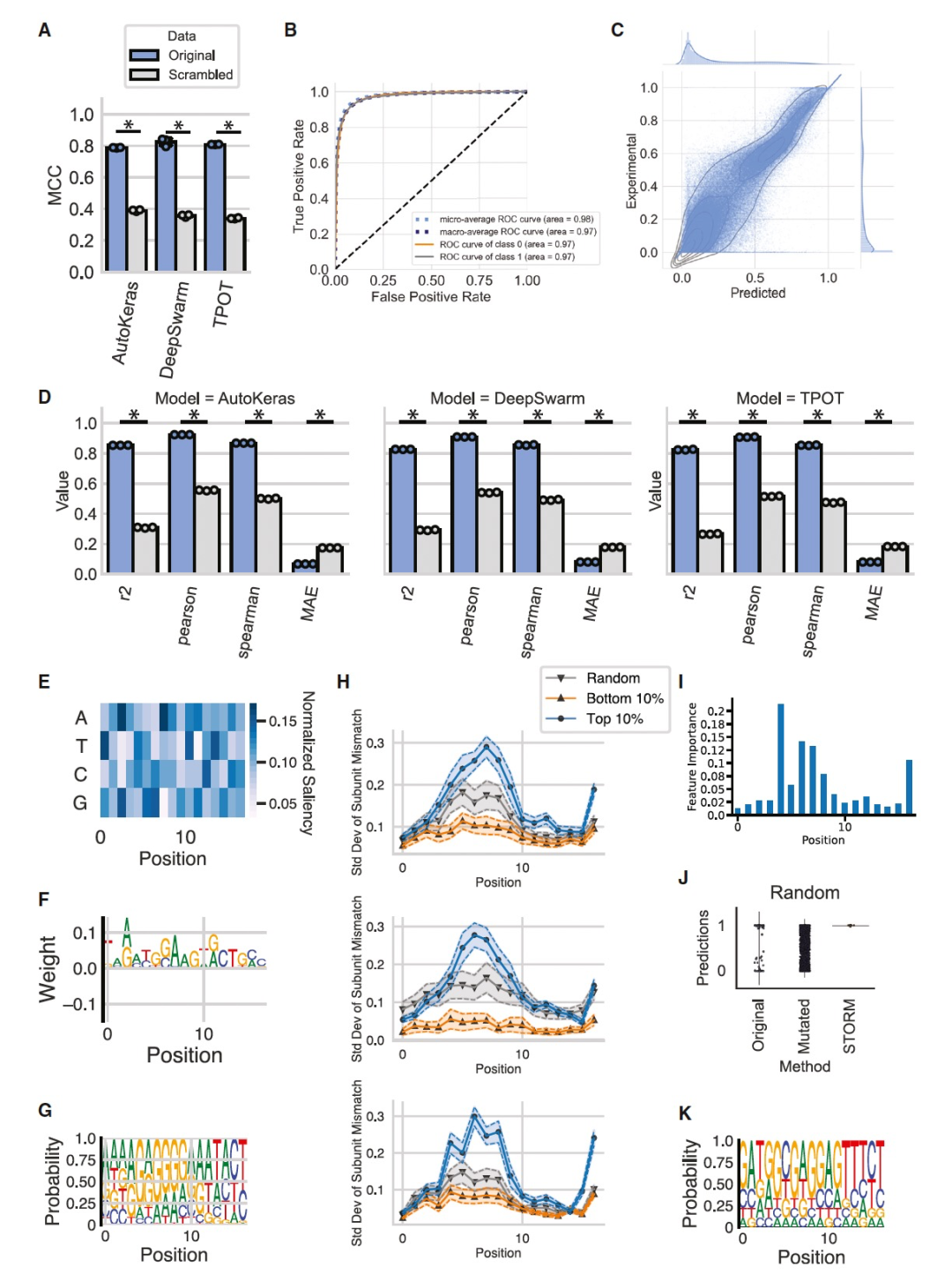
图 2
作者使用集成的AutoML框架进行了一系列案例研究。首先,作者探索了基因调控元素序列与效果之间的关系,具体而言,研究了RBS序列对大肠杆菌中翻译效率的影响。Ho¨ llerer等人使用基于DNA的表型记录技术,利用位点特异性重组酶研究了276,000个不同的17核苷酸长的RBS序列的翻译动力学。作者将BioAutoMATED应用于这个数据集,仅从序列中预测翻译效率。BioAutoMATED很容易生成具有Matthews相关系数(MCC) 0.8的二元分类模型,适用于所有三种搜索算法(Figure 2A)。最具预测性的模型是由DeepSwarm算法识别和训练的,其auROC为0.971,MCC为0.825 (Figure 2B)。在类似的回归任务中,BioAutoMATED使用AutoKeras搜索方法为RBS数据集确定了模型架构,其平均Pearson R为0.924,平均R2为0.854 (Figures 2C和2D),与Ho¨ llerer等人手动定义和调整的模型报告相符。他们使用残差网络(ResNet42)架构实现了R2在0.8和0.95之间的模型。作者将该回归模型在与Ho¨ llerer等人相同的RBS序列测试集上进行了测试,取得了类似的性能表现,auROC为0.939,Pearson R为0.931,R2为0.867,而Ho¨ llerer等人的R2为0.927 (表S1)。尽管BioAutoMATED的性能略低于最佳报告模型,但最佳性能的DeepSwarm回归模型仅用了26.5分钟 (Figures S2A和S2E),并且仅需十行用户输入(而Ho¨ llerer等人的模型则需超过750行代码)。此外,Ho¨ llerer等人的模型还需要手动选择架构和超参数,并使用随机网格搜索进行优化,这是一个需要一定ML专业知识的过程。而BioAutoMATED不受这些要求的限制。
为了更好地理解模型的预测结果和序列模体对翻译效率的影响,作者进行了一个控制实验,将输入序列进行乱序处理。BioAutoMATED可以自动执行这个实验,并通过对核苷酸顺序进行随机排列生成乱序序列。由此产生的数据集具有相同数量的序列,可以用来训练一个只基于核苷酸频率预测翻译效率的模型。在使用这个乱序控制的RBS模型进行评估时,可以发现这些模型在仅考虑核苷酸组成的情况下在RBS序列上表现显著较差(Figures 2A和2D)。这些结果定量地证实了RBS序列顺序对翻译效率的重要性,并与翻译起始模型相一致。作者进一步探究了RBS中是否有特别重要的模体或区域。BioAutoMATED的可解释性工具能够回答这个问题。作者从显著性图生成了RBS序列的logo图(Figures 2E和2F),发现其中包含AGATGG和TGGAAG模体,这与Ho¨ llerer等人报道的Shine-Dalgarno样模体(“AGGAGG和其亚序列”)相似。这个模体还可以与最佳实验序列的序列logo进行对比(Figures 2G),其中包含“AGAGGG”序列和大量的A和G核苷酸。虽然显著性图中的模体与背景相比仅略有丰度,但该模体出现在序列中间,大约在5-8个位置:这些位置被发现在预测的模型分数变化最大的位置(Figure 2H)和最高的特征重要性位置(Figure 2I)。
BioAutoMATED使得药物结合抗体序列的优化成为可能
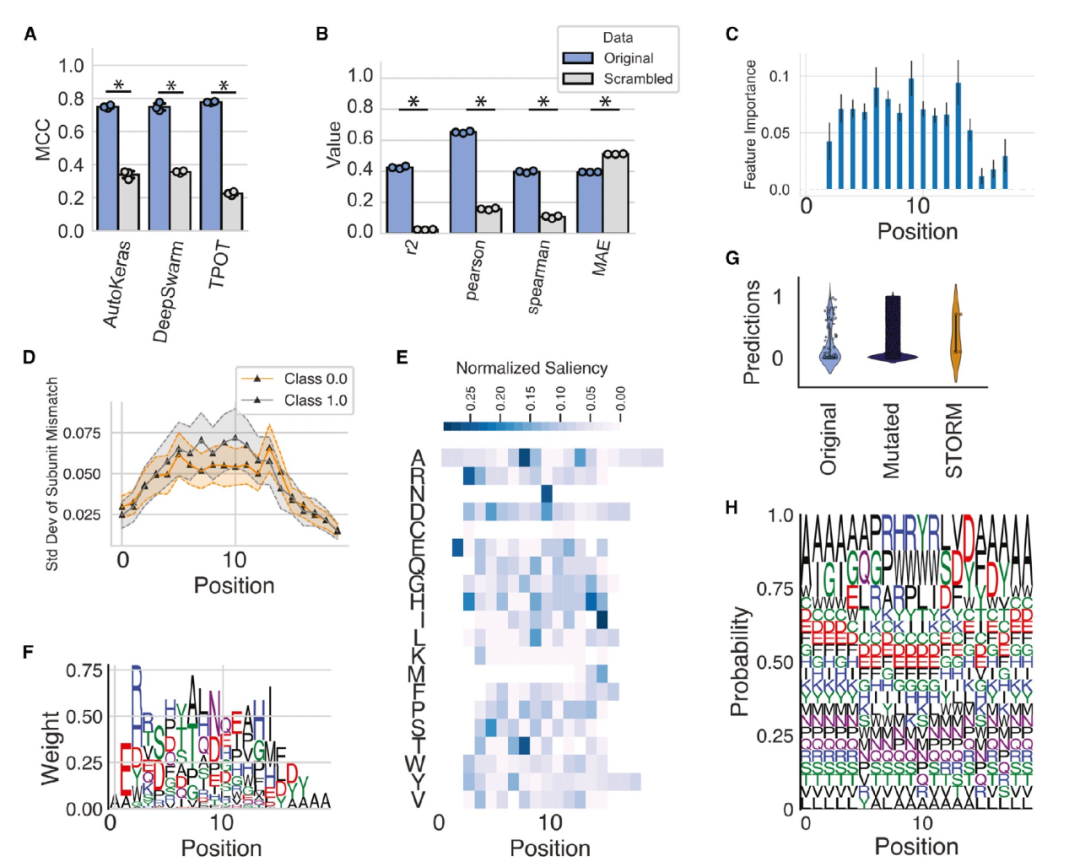
图 3
作者接下来研究了人类IgG抗体的精准靶向,探讨抗体序列在CDRH3区域的变化如何影响其与药物Ranibizumab的结合亲和力。作者将BioAutoMATED平台应用于一个包含67,769个肽序列的数据集,其中目标值对应于这些肽序列与Ranibizumab的结合富集情况。在该研究中,使用噬菌体展示技术对成千上万个抗体片段序列进行了结合亲和力的测定,旨在通过序列来预测抗体亲和力,并利用这些训练好的模型来设计新的抗体。
与RBS数据集类似,BioAutoMATED找到了高准确性的模型架构,这些模型的预测能力显著优于使用打乱顺序的肽序列训练的模型(图3A)。作者的最佳模型具有较高的预测能力,auROC为0.880,MCC为0.748,表明可以通过序列准确预测抗体亲和力,图3B。在这里,BioAutoMATED模型的令人鼓舞的性能能够生成预测模型并解决与解释性和设计相关的问题。为了研究解释性,作者询问了哪些氨基酸对预测的抗体亲和力与Ranibizumab的结合特别重要。通过生成特征重要性图(图3C)和in silico突变图(图3D),可以发现序列的前两个和后两个位置明显对结合亲和力的重要性贡献较小。这一结论在saliency map中得到了进一步支持(图3E和3F)。更详细地检查saliency map序列标志(图3F)以及通过序列标志可视化的性能最佳的肽序列,可以发现在15-17位置存在一个FDY模体,其中这一模体被发现对应于性能最佳的序列。saliency map还揭示了氨基酸的降低重要性,如甲硫氨酸、半胱氨酸和天冬氨酸,在整个序列中几乎没有相关的重要性值。
BioAutoMATED准确地根据功能对糖类进行分类
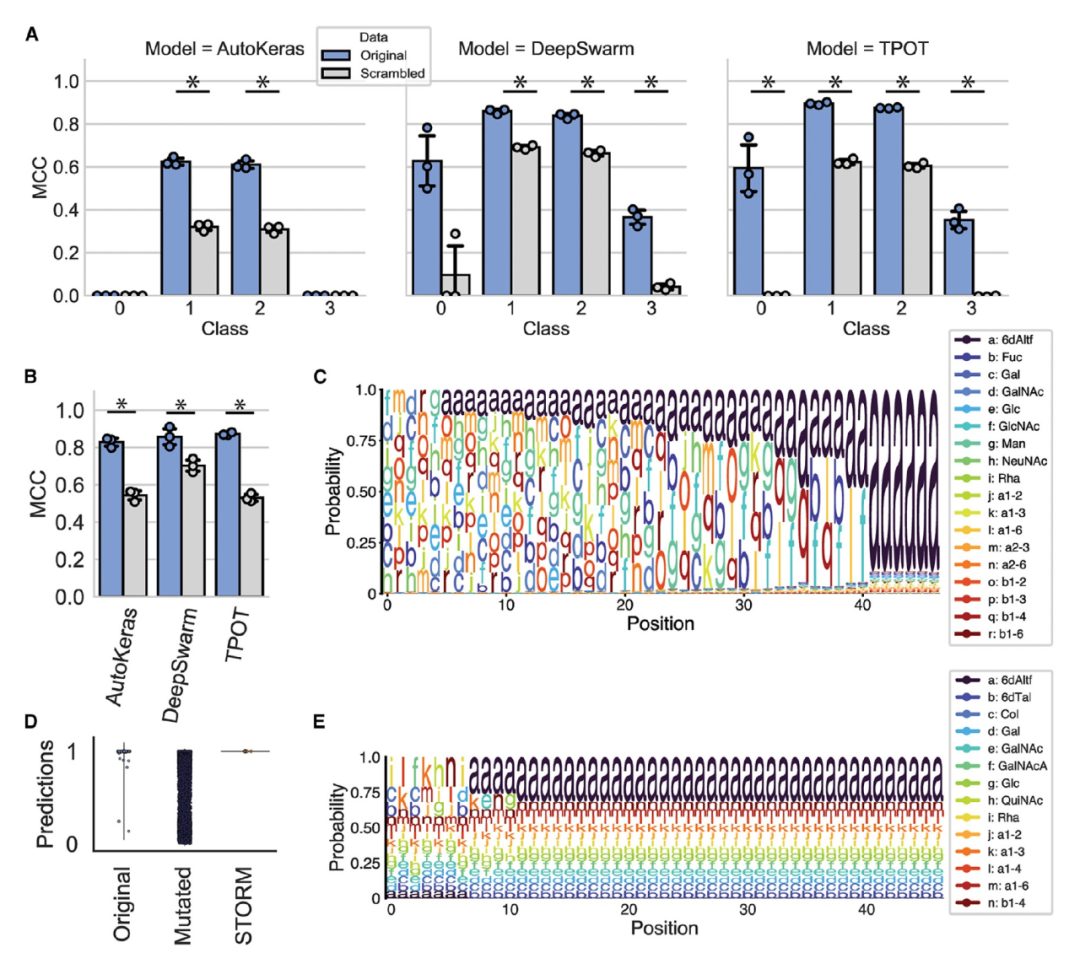
图 4
为了展示BioAutoMATED如何应用于更复杂的搜索空间,作者使用BioAutoMATED准确地对糖类进行分类。糖类是由单糖以特定排列方式形成的大型碳水化合物生物聚合物,具有广泛的分支结构,在宿主-病原体相互作用、细胞间通信、细胞粘附、自免疫和癌症等方面发挥着重要作用。最近在糖生物学中使用机器学习的成功,加强了将计算分析应用于不断增长的糖类序列数据库的兴趣。在这里,作者基于Bojar等人的工作,提供了一个经过精心筛选的糖类数据库‘‘SugarBase’’,并使用这个数据集来探索糖类序列组成对生物分类群和对人类的免疫原性的影响。这些糖类序列由单糖(例如半乳糖)和键(例如a-1,3键)组成,使用糖类的标准符号命名法表示。
首先作者使用BioAutoMATED进行多类分类,对生物分类群进行分类。基于序列进行分类的生物分类群是一个开放性问题,与微生物组样本分析相关,因为许多数据集缺乏适当的分类注释。BioAutoMATED模型表现良好(图4A),最佳性能的TPOT模型对古菌(34个序列)、细菌(5,856个序列)、真核生物(6,635个序列)和病毒(149个序列)领域分别实现了MCC值为0.594、0.895、0.875和0.351。这些结果与Bojar等人的领域MCC值(0.869)相当,并且显著高于对应的随机对照值,我们发现随机对照值分别为0.0、0.623、0.605和-0.002。
其次,作者使用BioAutoMATED对来自Bojar等人的1,320个标记的免疫原性或非免疫原性糖类序列进行二元分类。我们发现BioAutoMATED模型对这些序列进行训练后预测性能非常好,最佳的TPOT模型实现了auROC值为0.936、MCC值为0.873和准确率为92.7%(图4B)。这些模型与Bojar等人的最佳语言模型相当,其准确率为92%。使用TPOT模型评估的自动生成的随机对照模型实现了auROC值为0.763和MCC值为0.526,与非随机化模型相比有显著降低,但比Bojar等人使用随机糖类序列训练的语言模型更具预测性(准确率为51%)。Bojar等人报告称,他们的控制模型将糖类序列视为非语言(类似于BioAutoMATED),由于糖类的顺序和模式的功能重要性,准确率在80%至88%之间。保留糖类作为序列而不将其传递给语言模型,可能有助于从单糖和键组成中学习。
结论
这项工作介绍和评估了BioAutoMATED,一个用于集成和部署生物学序列研究的AutoML工具的平台。与其他AutoML方法相比,BioAutoMATED提供了几个独特的特性:(1) 它支持特定于序列的数据预处理,纠正了序列长度的变化;(2) 它处理糖类序列,除了核苷酸和蛋白质序列;(3) 它通过基于梯度上升的定向设计模块实现序列设计。此外,BioAutoMATED适用于各种任务,并提供了全面的解释模块,用于研究模型。BioAutoMATED在计算和生物领域平衡了最小的用户输入和多种自由度,从二元分类的阈值到选择模型解释的激活图梯度。在基本用户输入和数据上传后,BioAutoMATED可以自动进行下一步操作。当架构搜索结束时,平台提供的Python笔记本使用户能够在不与底层模型生成代码交互的情况下继续探索BioAutoMATED的功能。
参考资料
Valeri JA, Soenksen LR, Collins KM, Ramesh P, Cai G, Powers R, Angenent-Mari NM, Camacho DM, Wong F, Lu TK, Collins JJ. BioAutoMATED: An end-to-end automated machine learning tool for explanation and design of biological sequences. Cell Syst. 2023 Jun 21;14(6):525-542.e9. doi: 10.1016/j.cels.2023.05.007. PMID: 37348466.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-09-13 00:01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号