超级增强子系列11: 邪修GSEA-超级增强与peak集的富集分析R代码分享(建议收藏 )

超级增强子系列11: 邪修GSEA-超级增强与peak集的富集分析R代码分享(建议收藏 )

三兔测序学社
发布于 2025-12-29 10:07:09
发布于 2025-12-29 10:07:09
前言
GSEA的核心原理: GSEA(Gene set enrichment analysis,基因集富集分析)通过将全基因组基因按表达差异程度排序,计算预定义基因集在排序列表顶/底部的富集程度。其统计基础基于置换检验法,通过计算富集分数(ES)衡量基因集与表型相关性,最终以标准化富集分数(NES)和错误发现率(FDR)评估显著性。
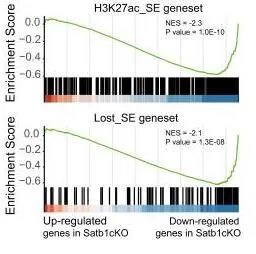
常规应用:在先前发表的超级增强子文章中,建立了两个SE相关基因集。用Satb1cKO 与WT DP细胞的RNA-seq数据构建基因排序文件:全基因的log2FC(cKO vs WT)文件。然后分析这两个基因集富集在哪一端。如图所示:这两个基因集均在Satb1cKO 细胞的下调基因中富集。说明Satb1的敲除会影响超级增强子相关基因的表达。
代码:常规的基因集富集分析
# 安装必要的包(首次运行时取消注释)
# BiocManager::install(c("clusterProfiler", "org.Hs.eg.db", "enrichplot", "DOSE"))
# install.packages("dplyr") # 用于数据处理
# 加载库
library(clusterProfiler)
library(org.Hs.eg.db) # 人类基因注释库,其他物种请更换
library(enrichplot)
library(dplyr)
library(DOSE)
# 设置工作目录并读取数据
# 假设你的数据有两列:SYMBOL, logFC
setwd("你的工作目录路径")
deg_data <- read.table("genelog2fC.txt", header = TRUE, sep='\t', stringsAsFactors = FALSE)
# 查看数据前几行,确保读取正确
head(deg_data)
# --- 步骤 A:基因 ID 转换 (Symbol 转 EntrezID) ---
# 注意:GSEA 对 ID 重复很敏感,这里进行去重处理
gene_mapping <- bitr(deg_data$SYMBOL,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
# 合并原始数据与转换后的 ID
merged_data <- inner_join(deg_data, gene_mapping, by = c("SYMBOL" = "SYMBOL"))
# --- 步骤 2:去重处理 (核心修改点) ---
# 使用 dplyr 对 SYMBOL 进行分组,并保留每组中 |log2FC| 最大的那一行
# 这样确保了每个 SYMBOL (以及对应的 ENTREZID) 只出现一次
merged_data_unique <- merged_data[!duplicated(merged_data$SYMBOL), ]
# 3. 确认行数是否减少
nrow(merged_data)
nrow(merged_data_unique)
# --- 步骤 B:构建 geneList ---
# 提取 logFC 作为排序依据,并设置名字为 ENTREZID
geneList <- merged_data_unique$log2fc
names(geneList) <- merged_data_unique$SYMBOL
# 按 logFC 从大到小排序 (这是 GSEA 的核心输入)
geneList <- sort(geneList, decreasing = TRUE)
# 读取自定义 GMT 文件
# 格式要求:第一列是通路 ID,第二列是描述(可选),后面全是基因(ENTREZID 或 SYMBOL)
# 注意:如果 GMT 中是 Symbol,上面的 geneList 名字也必须是 Symbol;推荐统一用 EntrezID。
custom_gmt <- read.gmt("lost_gained_SEgeneGMT.gmt")
# 查看前几行结构
head(custom_gmt)
# 在运行 GSEA 前加上这行
set.seed(123)
# 执行 GSEA 分析
gsea_result <- GSEA(geneList = geneList,
TERM2GENE = custom_gmt, # 传入自定义基因集
minGSSize = 50, # 最小基因集包含基因数
maxGSSize = 1000, # 最大基因集包含基因数
pvalueCutoff = 1, # P值阈值 (GSEA通常先看NES,不严格过滤)
verbose = FALSE,
seed = TRUE)
# 查看结果
head(gsea_result@result)
write.csv(gsea_result@result, "SE_GSEA_results.csv", row.names = FALSE)
# 获取排名靠前的通路 ID,如果是自己自定义基因集,可以展示全部。
top_terms <- gsea_result@result$ID[1:2]
# 绘制图形
p1 <- gseaplot2(gsea_result,
geneSetID = top_terms, # 传入前2个ID的向量
title = Lost_Gained_SE-Gene_GSEA",
color = c("#4DBBD5", "#E64B35"), # 可以指定单一颜色,也可以传入颜色向量 c("red", "blue")
base_size = 15,
pvalue_table = TRUE) # 显示包含 Pvalue 等信息的表格
# 显示图形
print(p1)邪修GSEA:特征集的富集分析(Feature set enrichment analysis:FSEA)
当我们了解GSEA基本原理就知道,其实就是一个特定基因集合 在排序的整体基因集合中富集方向的判断。如果我们将基因名称,换成其他特征如DNA上的Peak位置、蛋白名称,代谢产物、微生物名称等。都是可以的。只要a 子集在ALL 集合中,并且ALL中每一个都有独特的ID号与排序位置,就可以进行特征集的富集分析(Feature set enrichment analysis).
差异超级增强子区域的染色质可及性或TF结合富集分析
分析流程举例说明:
1.在肿瘤细胞与健康细胞中进行了H3K27ac ChIP-seq分析,进行了超级增强子的鉴定,并分析了在肿瘤细胞中丢失或者获得性的增强子区域(lost/gained SE)
2.在肿瘤细胞与健康细胞中进行了ATAC-seq 或者 TF ChIP-seq,并进行了差异分析。获得peaks 位置与对应log2FC值(cancer vs normal).
2.1 构建peakID.bed。在Excel或WPS表格中:使用公式 =A2&"-"&B2&"-"&C2(假设chr, start, end分别在A、B、C列),然后向下填充。在R语言中:使用 paste 函数,例如 dfstart, dfend, sep="-")。
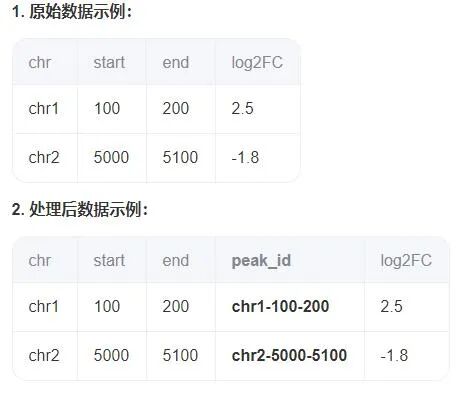
2.2 获得PeakIDlog2FC.txt文件用于GSEA分析。将peakID.bed文件的peak_id与log2FC列提取出来,并以PeakIDlog2FC.txt进行保存。
3.构建Lost/Gained SE相关的ATAC-seq peaks或TF ChIP-seq peaks的GMT文件(特征集)
bedtools intersect -a peakID.bed -b Lost_SE.bed Gained_SE.bed -filenames -wa -wb > lostgainedSE-ATACpeakset.bed 然后在Excel 或者WPS中, lostgainedSE-ATACpeakset.bed 文件,分别筛选出包含 “Lost_SE.bed” 和 “Gained_SE.bed” 的行。并将peak_id列复制新文件中。
lostgainedSE_featurePeak.gmt 文件内容示例:
Lost_SE_Peaks Lost_SE_Peaks chr1-100-200 chr3-500-600
Gained_SE_Peaks Gained_SE_Peaks chr2-5000-5100 chr5-8000-82004.确认两个文件:PeakIDlog2FC.txt,lostgainedSE_featurePeak.gmt
后续R代码如下
# 加载库
library(clusterProfiler)
library(org.Hs.eg.db) # 人类基因注释库,其他物种请更换
library(enrichplot)
library(dplyr)
library(DOSE)
library(ggplot2)
# 设置工作目录并读取数据
setwd("你的工作目录路径")
deg_data <- read.table("PeakIDlog2FC.txt", header = TRUE, sep='\t', stringsAsFactors = FALSE)
# 查看数据前几行,确保读取正确
head(deg_data)
# --- 步骤 2:去重处理 (核心修改点) ---
merged_data_unique <- deg_data[!duplicated(deg_data$peak_id), ]
# 3. 确认行数是否减少
nrow(deg_data)
nrow(merged_data_unique)
# --- 步骤 B:构建 geneList ---
# 提取 log2FC 作为排序依据
geneList <- merged_data_unique$log2FC
names(geneList) <- merged_data_unique$peak_id
# 按 logFC 从大到小排序 (这是 GSEA 的核心输入)
geneList <- sort(geneList, decreasing = TRUE)
custom_gmt <- read.gmt("lostgainedSE_featurePeak.gmt")
# 查看前几行结构
head(custom_gmt)
# 在运行 GSEA 前加上这行
set.seed(123)
# 执行 GSEA 分析
gsea_result <- GSEA(geneList = geneList,
TERM2GENE = custom_gmt, # 传入自定义特征集
minGSSize = 50, # 最小特征集包含peakID数量
maxGSSize = 1000, # 最大特征集包含peakID数量,可自由调整,大于自己要分析的相关集。
pvalueCutoff = 1,# P值阈值 (GSEA通常先看NES,不严格过滤)
verbose = FALSE,
seed = TRUE)
# 查看结果
head(gsea_result@result)
write.csv(gsea_result@result, "GSEA_results.csv", row.names = FALSE)
#获取前2个Term 的 ID
top_terms <- gsea_result@result$ID[1:2]
# 绘制图形
p1 <- gseaplot2(gsea_result,
geneSetID = top_terms, # 传入前2个ID的向量
title = "差异增强子与可及性或转录因子富集差异",
color = c("#4DBBD5", "#E64B35"), # 可以指定单一颜色,也可以传入颜色向量 c("red", "blue")
base_size = 15,
pvalue_table = TRUE) # 显示包含 Pvalue 等信息的表格,无法显示NES。
# 显示图形
print(p1)
# 保存为PDF格式
ggsave(filename = "GSEA_Plot.pdf",
plot = p1,
device = "pdf",
width = 10, # 可根据需要调整宽度(单位:英寸)
height = 8, # 可根据需要调整高度(单位:英寸)
dpi = 300) # 分辨率
# 保存为EPS格式(矢量图,适合编辑)
ggsave(filename = "GSEA_Plot.eps",
plot = p1,
device = "eps",
width = 10,
height = 8)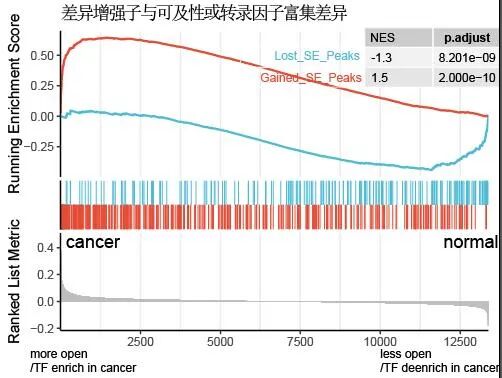
此图说明:获得性的超级增强子所在的染色质可及性区域或者TF(p300)结合在肿瘤中(与健康细胞比较)更开放或者更富集。而丢失的SE区域的染色质可及性在肿瘤细胞中降低,TF结合也降低,在健康细胞中富集更明显。
科研不止于工具,更在于思路。选择正确的工具,才能让数据说话。
【超级增强子系列文章】
超级增强子系列1:super enhancer鉴定-ROSE软件的安装与使用
超级增强子系列2:ROSE准备gff文件:peak 信息文件转化为9列gff格式文件R代码
超级增强子系列3:R语言批量处理ROSE文件生成SE与TE.bed文件
超级增强子系列5:用ChIPseeker进行超级增强子基因注释
超级增强子系列6:GREAT-基因组调控元件专业注释富集工具
超级增强子系列7: 用MEME进行超级增强子转录因子motif 富集分析实战
超级增强子系列8: motif 富集分析工具XSTREME输出文件解释
超级增强子系列9: ggseqlogo进行Motif文件可视化
超级增强子系列10: bedtools筛选Lost/Gained超级增强子
如果你觉得这篇博文对你有帮助,请点赞、收藏、转发!支持我们持续输出优质内容!
关注“三兔测序学社”,获取更多实用教程与前沿解读。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-12-25,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号