基于 germline 信息进行样本配对性检验


简介
在肿瘤基因组测序数据分析的过程中,一般要求需要肿瘤和正常样本配对。然而在样本收集、实验处理、送样测序等过程中,由于实验人员的失误,可能存在样本混淆的情况,即肿瘤A和正常A可能不是来自于病人A。但对于分析人员来说,要重新对样本进行溯源是难以实现的操作。那么如何从分析方法上来判断肿瘤和正常是否配对或者是否来自于同一个病人?
问题描述
一般肿瘤基因组数据分析,都会对体细胞突变结果进行突变全景图或者瀑布图可视化,如下示例:
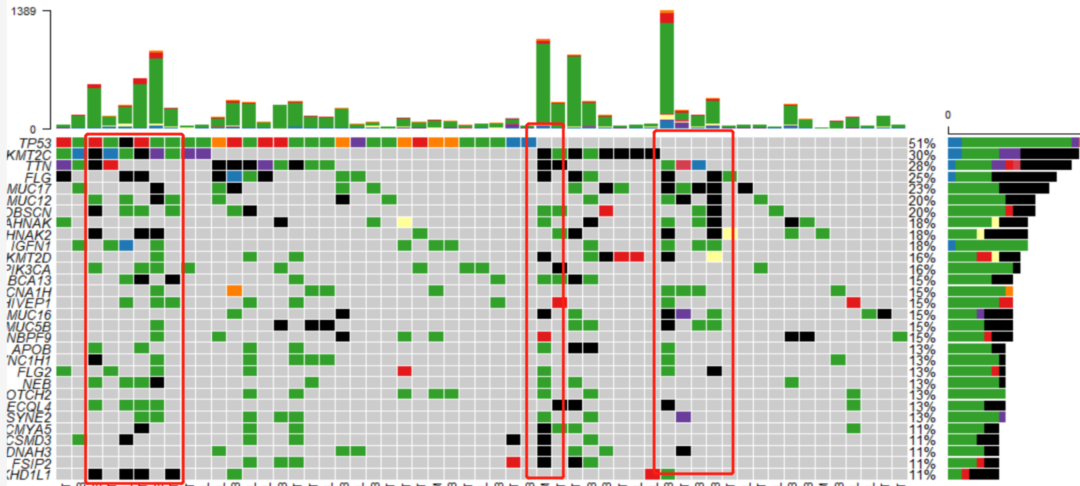
如果一批病人的肿瘤和正常样本中,有某对肿瘤-正常样本并非来自于同一个病人,在突变瀑布中的表现就是这个肿瘤样本的体细胞突变数量较多。Multi_hit(即同一样本的同一个基因出现多次突变)图中黑色方块较多,当然也不能排除某些样本存在超突变的情况。但一般出现这个问题,就建议对这一批样本进行样本配对性检验。
方法实现
可以使用现成的工具,如NGScheckmate:https://github.com/parklab/NGSCheckMate
但也可以对样本的 SNP基因分型进行一致性分析。即肿瘤或者正常样本的 germline 信息中含有 SNP 分型,一般来自于同一个病人的样本,其SNP 分型基本一致。一个简单方法是同一个批病人的多个样本,基于 SNP 分型信息进行聚类,即可以判断样本是否来自于同一病人。
实现过程是,对同一批病人的肿瘤和正常样本,检测 germline 突变,可以用 GATK 的 HaplotypeCaller GVCF 模式,最后得到的为多样本合并后的 vcf 文件。其中 Format 信息中的 GT tag 记录了样本的基因型信息,从 vcf 文件中的格式说明可以看到:
GT : genotype, encoded as allele values separated by either of / or |. The allele values are
- 0 for the reference allele (what is in the REF field),
- 1 for the first allele listed in ALT,
- 2 for the second allele list in ALT and so on.
For diploid calls examples could be 0/1, 1|0, or 1/2, etc. For haploid calls, e.g. on Y, male nonpseudoautosomal X, or mitochondrion, only one allele value should be given; a triploid call might look like 0/0/1. If a call cannot be made for a sample at a given locus, '.' should be specied for each missing allele in the GT field (for example './.' for a diploid genotype and '.' for haploid genotype). The meanings of the separators are as follows (see the PS field below for more details on incorporating phasing information into the genotypes):
- / : genotype unphased
- | : genotype phased
可以从多样本的 vcf 文件中将这部分信息提取出来,进行 SNP 指纹聚类。如果将整个vcf文件的所有 snp 位点进行聚类的话,数据量较大,可以提取某一部分的 snp 位点进行聚类即可。
多样本的 germline VCF 示例如下:第11列开始是样本对应的 FORMAT 信息。
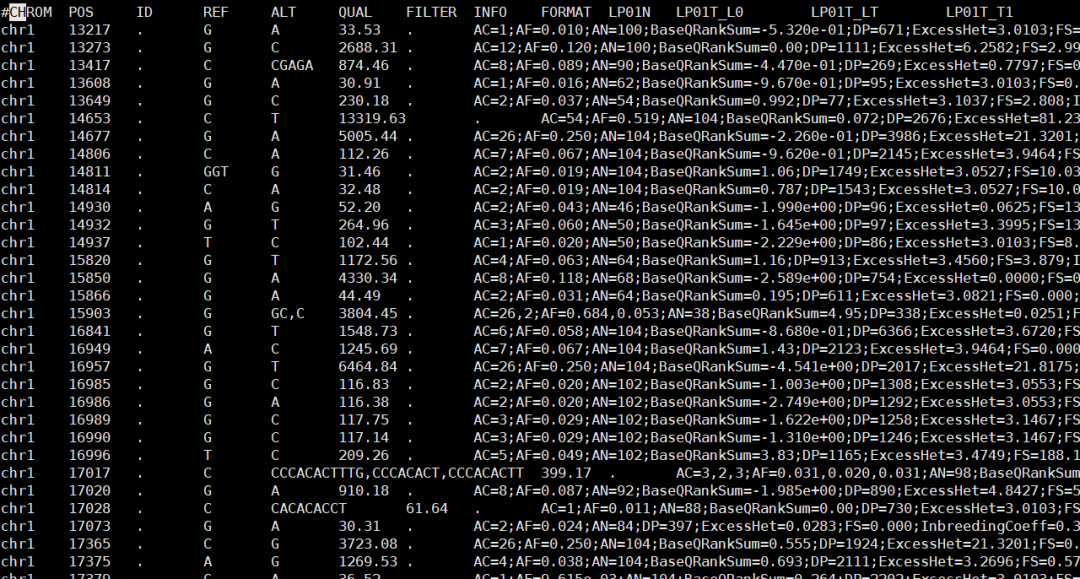
如这里对一个多样本的 vcf 文件 germline_merge.vcf.gz 提取 chr1 的 vcf 信息:
zcat germline_merge.vcf.gz | grep -v '##'| \awk 'BEGIN{FS="\t";OFS="\t"} \- {if(NR==1) {print 0} else if(1=="chr1" && 7=="PASS") print
gzip >chr1.vcf.gz
然后基于R语言进行数据处理和聚类可视化:
rm(list = ls())library(tidyr)library(data.table)library(stringr)library(pheatmap)vcf = fread("chr1.vcf.gz")vcf =as.data.frame(vcf)colnames(vcf)[1]="CHROM"vcf = unite(vcf,"Mut_site","CHROM","POS")vcf = vcf[,-c(2:8)]rownames(vcf)= vcf[,1]vcf = vcf[,-1]# 通常 GT 在 format 的第一个tag,所以可以去除掉其他 tagfor(i in1:ncol(vcf)){vcf[,i]= gsub(":.*","",vcf[,i])}# 因为测序不是所有位点都有覆盖,所以将一半样本没有覆盖到的突变位点过掉掉re = c()for(i in1:nrow(vcf)){re1 = ifelse(sum(vcf[i,]=="./.")> ncol(vcf)/2,F,T)re = c(re,re1)}vcf = vcf[re,]# 如果 GT 为 0/1 0|1 1/1 1|1 即 纯合突变和杂合突变,都赋值为 1for(i in1:ncol(vcf)){vcf[,i]= ifelse(str_detect(vcf[,i],"1"),1,0)}# snp = fread('snp.01.txt')# colnames(snp)# snp = tidyr::unite(snp, "variant", sep = ":", CHROM, POS)# rownames(snp) = snp$variant# snp=snp[,-1]snp = t(vcf)png(filename ='snp.png',width =1080,height =1080)pheatmap::pheatmap(snp,cluster_rows = T,cluster_cols = F,annotation_names_row = T,show_rownames = T,show_colnames = F)dev.off()
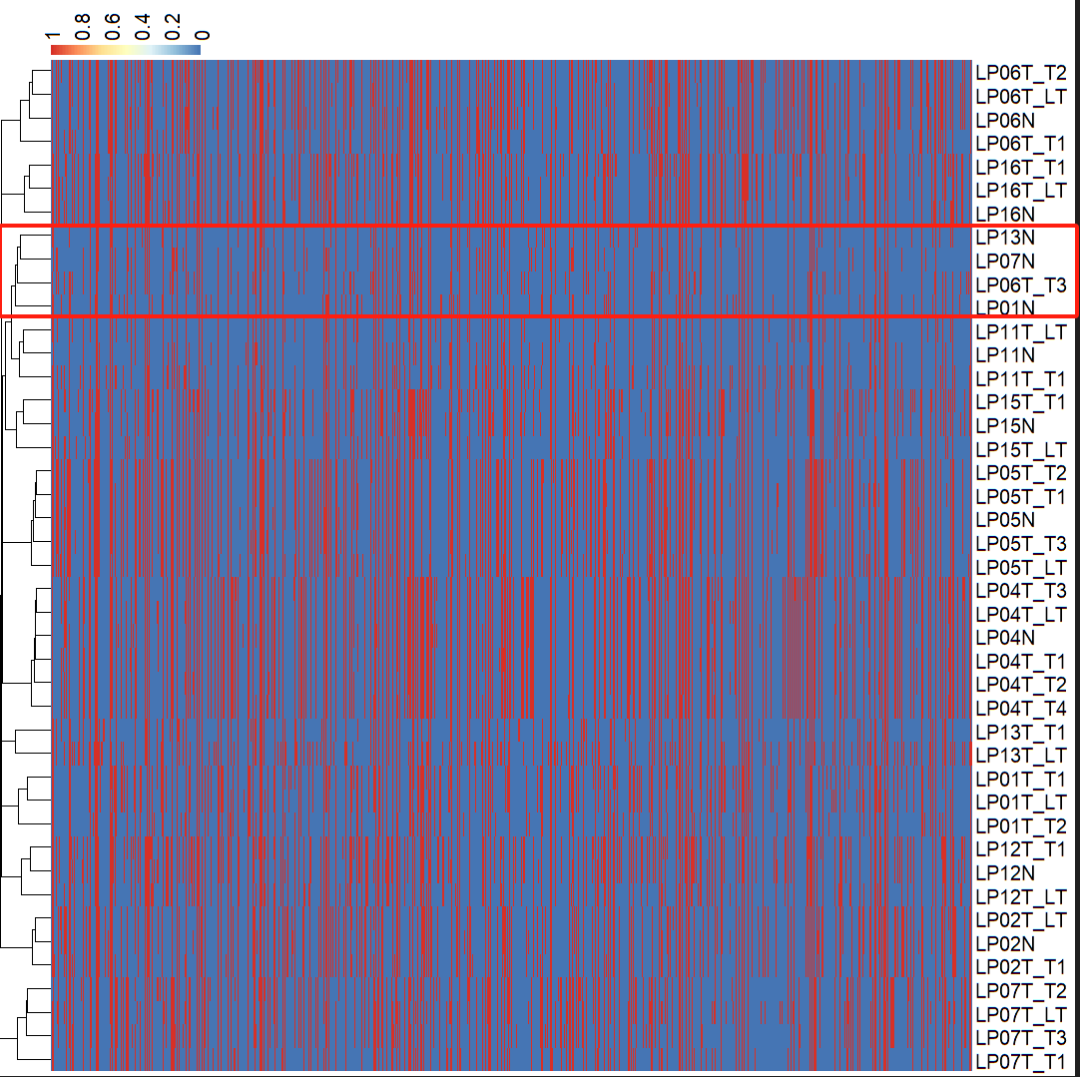
从可视化的结果上看,病人01、06、07、13 的肿瘤和正常样本就没有聚类到一起,如果这几个病人的肿瘤样本体细胞突变数较多,则有证据怀疑其成对样本可能混淆。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-07-24,如有侵权请联系 cloudcommunity@tencent.com 删除
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号