The Innovation Informatics|清华团队提出 MolF-DAEs,将 190 万活性分子压缩进三维化学空间

The Innovation Informatics|清华团队提出 MolF-DAEs,将 190 万活性分子压缩进三维化学空间

MindDance
发布于 2026-05-20 13:38:19
发布于 2026-05-20 13:38:19
过去几年,人工智能正在快速进入药物发现流程。
从分子性质预测、虚拟筛选,到生成式分子设计,越来越多模型开始帮助研究人员在庞大的化学空间中寻找潜在候选分子。然而,对于药物发现而言,仅仅预测某个分子的活性还不够。更关键的问题是:如何理解这些分子在化学空间中的组织方式?哪些分子因为相似的子结构而聚在一起?这些聚类又是否对应相似的生物活性?
这并不是一个简单的可视化问题。
生物活性分子通常由高维、稀疏的分子指纹表示。传统方法往往需要先用 PCA、UMAP 或 t-SNE 等降维方法把高维数据投影到二维或三维空间,再进行聚类和可视化。但这种两步流程可能引入几何扭曲,使原本存在于高维空间中的结构—活性关系变得模糊。
近期,来自清华大学深圳国际研究生院的谭英、陈宇综团队在 The Innovation Informatics 发表论文,提出了一种基于分子指纹的深度自编码器框架 MolF-DAEs,用于直接在三维潜在空间中分析 190 万级生物活性分子。

这项研究尝试回答一个核心问题:
能否在不依赖额外降维步骤的情况下,把高维分子指纹直接压缩到三维空间,同时保留分子结构、局部邻域关系和生物活性组织?
不是简单降维,而是端到端学习三维化学空间
研究团队提出的 MolF-DAEs,全称为 Molecular Fingerprint Deep Autoencoders,即分子指纹深度自编码器。
简单来说,自编码器由两个部分组成:编码器负责把高维分子指纹压缩成低维表示,解码器则尝试从低维表示中重构原始分子指纹。若模型能够在极低维度下较好地重构原始指纹,说明这个低维空间仍然保留了足够的化学信息。
在这项研究中,团队并没有先把分子压缩到较高维空间,再用 UMAP 或 PCA 继续投影,而是直接让模型学习一个三维潜在空间。每个分子最终都被表示为一个三维坐标点,也就是可以被直接放入 3D 化学空间中观察。
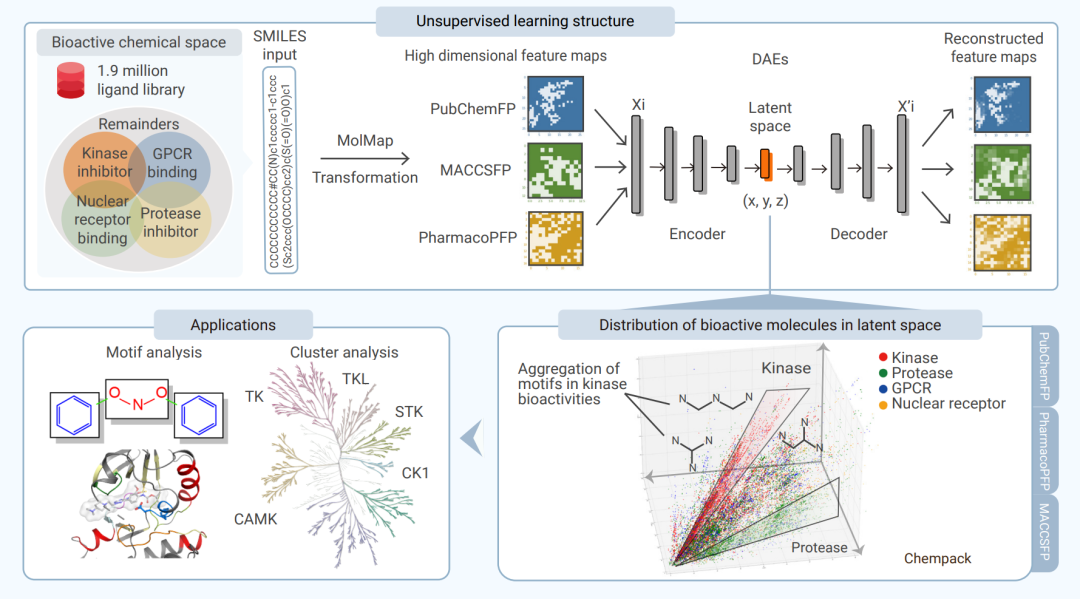
MolF-DAEs 用于生物活性分子无监督聚类、三维可视化和下游分析的整体流程。
MolF-DAEs 用于生物活性分子无监督聚类、三维可视化和下游分析的整体流程。
190 万个活性分子,三套分子指纹
为了构建这一三维化学空间,研究团队从 ChEMBL 数据库中收集了 1,937,109 个具有中高活性的生物活性分子。这些分子具有实验测得的 IC50、EC50 或 Ki 等活性指标,并覆盖从临床前到已获批阶段的化合物。
研究还标注了四类重要药物靶点相关分子,包括激酶、蛋白酶、GPCR 和核受体。其中,激酶相关分子约 12.46 万个,蛋白酶相关分子约 9.20 万个,GPCR 相关分子约 3.37 万个,核受体相关分子约 2.82 万个,其余 165 万余个分子被归为其他类别。
在分子表示方面,团队使用了三类常见分子指纹:PubChemFP、MACCSFP 和 PharmacoPFP。前两者更强调分子子结构特征,后者则更关注药效团的位置、空间关系和相互作用模式。
换句话说,MolF-DAEs 并不是只看一种分子描述方式,而是从不同指纹角度考察:高维化学信息能否被压缩进一个紧凑的三维空间。
三维压缩后,分子指纹仍能高精度重构
在极端压缩条件下,最直接的问题是:信息会不会丢失太多?
结果显示,MolF-DAEs 在三类分子指纹上都实现了较高的重构率。其中,PubChemFPM、MACCSFPM 和 PharmacoPFPM 的重构率分别达到 97.55%、96.10% 和 97.55%。这说明,即使每个分子最终只被压缩成三个坐标,模型仍然能够较好地保留原始分子指纹中的结构信息。
同时,研究团队还用 trustworthiness、KNN preservation、continuity 等指标评估局部邻域关系是否被保留。结果显示,MolF-DAEs 在不同分子指纹下均表现出较高的局部结构保真度,例如 PubChemFPM 的 trustworthiness 达到 0.955,PharmacoPFPM 达到 0.958。
这意味着,三维潜在空间中的“邻近分子”,在原始高维指纹空间中也大概率具有相近的结构特征。
活性分子在三维空间中形成“带状聚类”
更有意思的是,当 190 万个分子被投射到 3D 潜在空间后,研究团队观察到了一种特殊的空间结构:分子并不是随机分布,而是形成了从共同原点向外延伸的带状聚类。
这些带状结构在三套分子指纹中都可以观察到。每条带内部存在连续变化,同时又会出现一些相对独立的“岛状区域”。研究认为,这种结构反映了分子特征空间中的内在梯度,而不只是某种可视化算法造成的图形效果。
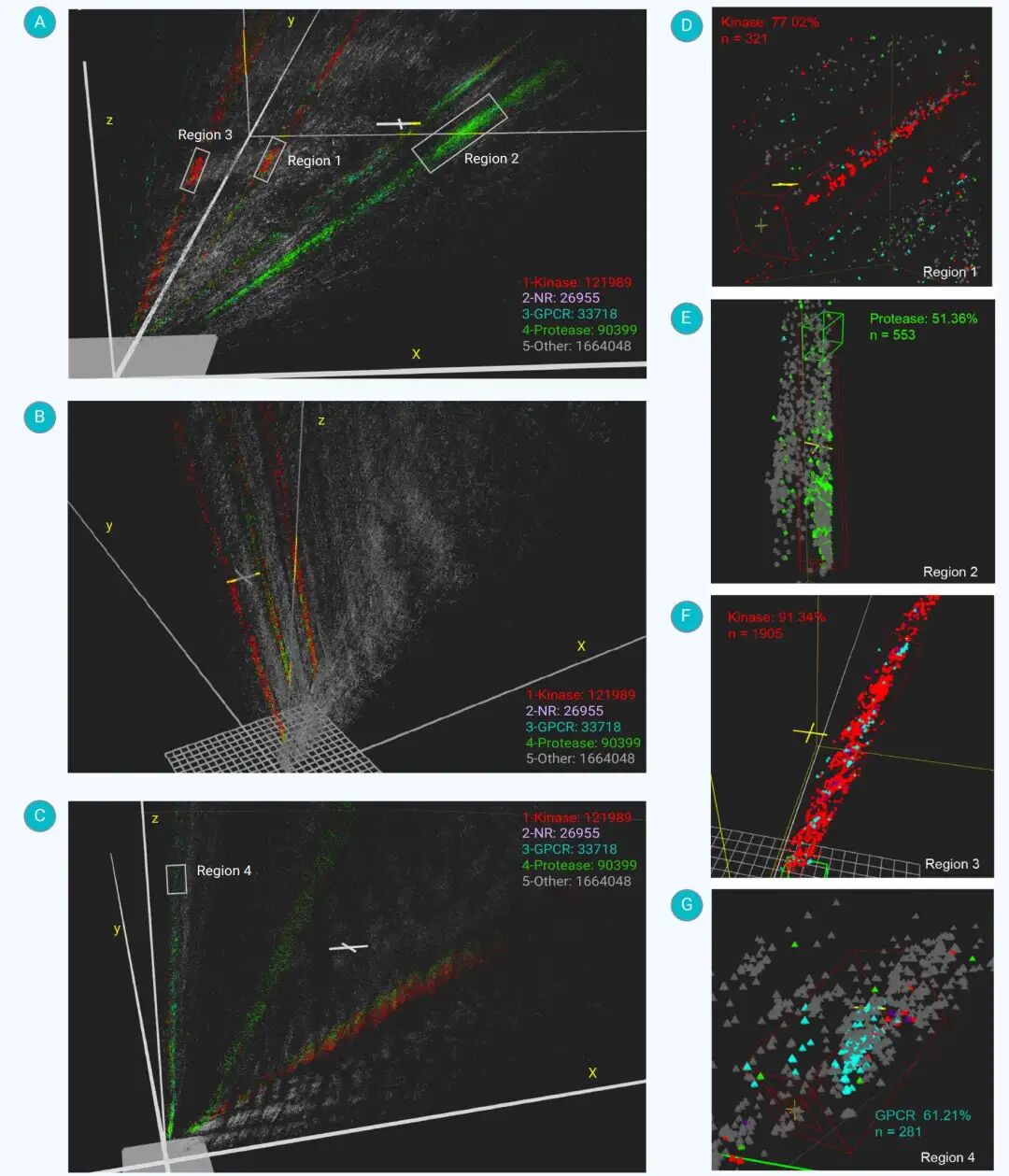
Chempack 软件展示的 190 万生物活性分子三维潜在空间分布,不同颜色对应不同靶点类型。
Chempack 软件展示的 190 万生物活性分子三维潜在空间分布,不同颜色对应不同靶点类型。
值得注意的是,MolF-DAEs 在训练时并没有使用靶点标签作为监督信息,但激酶、蛋白酶、GPCR 等靶点相关分子仍然在三维空间中自然呈现相对集中的区域。在代表性局部区域中,kinase、protease 和 GPCR 分子均出现明显富集,说明模型在无监督条件下捕捉到了与生物活性相关的分子组织规律。
这说明,模型并不只是完成了“好看的降维”,而是在无监督条件下捕捉到了与生物活性相关的分子组织规律。
每一条“带”,都对应特定的子结构特征
为什么这些分子会形成带状聚类?
研究团队进一步发现,不同带状区域往往由不同的核心子结构或药效团特征主导。也就是说,三维空间中的聚类并不是单纯由几何距离决定,而是与分子的结构片段和潜在生物活性有关。
以激酶相关分子为例,研究观察到多类激酶富集带,其中一些区域富含疏水环结构和核心氢键位点。这些结构元素与已知激酶抑制剂中的关键药效团模式具有一致性。
在 PubChemFPM 的部分激酶富集区域中,线性碳氮子结构 NC-N-C 几乎出现在所有已知激酶抑制剂中;同时,大量分子还连接至少一个六元环或五元环。这些结构特征有助于解释为什么相似活性的分子会在三维潜在空间中靠近。
蛋白酶相关分子则形成了另一个相对分离的带状区域。该区域主要由拟肽类骨架及其变体组成,可能与天然底物模拟和水解稳定性有关。GPCR 相关分子则更分散,常常表现为短而碎片化的岛状聚类,反映了 GPCR 结合口袋和天然配体的多样性。
Chempack:让化学空间可以被快速导航
为了更直观地浏览这些三维分布,研究团队还开发了 Chempack 软件。
Chempack 可以用于快速导航和显示 DAE 生成的三维分子分布景观,支持在局部三维立方区域中查看分子点,并通过不同颜色区分激酶、蛋白酶、核受体、GPCR 和其他靶点类型。
这一步很重要。
对于大规模分子库而言,模型不应只是给出一个抽象的向量,而应该帮助研究人员“看见”化学空间:哪些分子聚在一起,哪些区域对应已知靶点,哪些灰色未标注分子可能具有潜在新活性。
从“预测分子活性”到“理解结构—活性景观”
相比传统监督学习模型,MolF-DAEs 的目标并不是直接预测某个分子是否作用于某个靶点。
它更像是一种无监督的化学空间地图构建方法:把高维、稀疏、难以直观看懂的分子指纹,压缩成一个可导航、可解释的三维空间,并在其中揭示子结构、靶点类别和生物活性之间的对应关系。
研究团队指出,这种表示可用于配体虚拟筛选、骨架新颖性评估、取代基多样性分析、潜在脱靶作用预测、多靶点交叉反应分析,以及药物再利用等任务。
当然,这一方法也并非没有限制。由于模型强制把高维分子信息压缩到固定三维空间中,虽然提升了可视化清晰度和解释性,但也可能降低对复杂、多尺度化学关系的分辨能力。此外,三维空间中的大尺度距离关系并不一定总能对应真实化学相似性,因此在解释全局空间距离时仍需谨慎。
小结
这项研究展示了一种新的思路:AI 不只是给分子打分,也可以帮助研究人员重新组织和理解庞大的药物化学空间。
通过 MolF-DAEs,研究团队将约 190 万个生物活性分子直接嵌入三维潜在空间,在不依赖额外降维流程的情况下发现了具有靶点相关性和子结构特征的带状聚类。论文总结认为,该方法能够在无监督条件下保留结构—活性关系,并揭示新的分子骨架模式。
从更广的角度看,这项工作回应了 AI 药物发现中的一个基础问题:面对超大规模、高维、稀疏的分子数据,模型不仅要“预测得准”,还要让研究人员理解化学空间为什么这样组织。
这可能会成为未来可解释药物发现中的一条重要路径。
参考文献
Ying H., Wu X., Qin C., et al. (2026). Molecular-substructure Deep Autoencoders cluster bioactive molecules into novel band-shaped substructure-distinguished bioactivity clusters in 3D latent space. The Innovation Informatics 2:100049. https://doi.org/10.59717/j.xinn-inform.2026.100049
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-20,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号