文献解读-爱沙尼亚人群肠道微生物群中古菌群落的分布特征

文献解读-爱沙尼亚人群肠道微生物群中古菌群落的分布特征

用户1075469
发布于 2026-03-25 12:30:44
发布于 2026-03-25 12:30:44
Hello,Hello,小伙伴们大家好! 今天给大家推荐一篇非常实用的宏基因组学研究文章:《Metagenome-assembled genomes of Estonian Microbiome cohort reveal novel species and their links with prevalent diseases》。需要特别说明的是,这项研究最初以预印本形式发布在 bioRxiv 上,随后经过同行评议,正式发表在 Nature 旗下期刊 Scientific Data。
该研究基于爱沙尼亚微生物组队列(Estonian Microbiome cohort),构建了一套较为完整的宏基因组分析流程,涵盖 宏基因组测序、基因组组装、binning、MAG 质量评估、物种聚类、物种丰度计算以及疾病关联分析 等关键环节。文章同时在 GitHub 上公开了完整分析脚本,具有较好的可参考性和可复现性。该研究整合了 MEGAHIT、MetaBAT、MaxBin、VAMB 和 DAS Tool 等多种组装与 binning 工具,实现了复杂肠道微生物群落中高质量 MAG(Metagenome-assembled genomes) 的重建,并建立了人群特异性的微生物参考数据库。
对于希望系统了解或学习宏基因组分析流程、MAG 重建策略以及微生物与疾病关联分析方法的研究者而言,这篇文章具有较好的学习和参考价值。
摘要
虽然微生物群在维持人体健康中具有重要作用,但其中的古菌组分仍缺乏系统研究。基于爱沙尼亚微生物组深度队列(EstMB-deep)的 1,878 份粪便宏基因组样本,研究者构建了人群特异性的微生物参考基因组资源,并进一步对古菌组分进行了专项挖掘。研究共重建 84,762 个宏基因组组装基因组(MAGs),代表 2,257 个物种,其中包括 353 个潜在新物种;另有 607 个物种 未被全球统一人类胃肠道基因组(UHGG)参考数据库收录,提示其可能具有爱沙尼亚人群特异性。
与此同时,针对古菌的重建分析共鉴定出 273 个古菌 MAG,代表 21 个物种和 144 个菌株,并建立了 “EstMB MAGdb Archaea-273” 古菌 MAG 资源集。进一步的关联分析表明,在 33 种常见疾病中,共检测到 15 种疾病与 44 个微生物信号存在显著关联,其中涉及 10 个潜在新物种 和 5 个未被 UHGG 收录的物种。总体而言,该研究表明,基于人群队列的 de novo 宏基因组组装不仅能够补充现有参考数据库、揭示被忽视的微生物多样性,还可为解析微生物组与常见疾病之间的关联及人群特异性差异提供重要资源。
数据代码
“EstMB MAGdb Archaea-273” 古菌 MAGs 集合的序列数据已上传至 European Nucleotide Archive(ENA) 数据库,研究登录号为 PRJEB81541;
“EstMB MAGdb Archaea-273” MAGs 集合的描述信息,包括质量参数、分类学注释和基因组特征,已公开发布于 Figshare:https://doi.org/10.6084/m9.figshare.30920087;
“Archaea ESTrep-21” MAGs 集合的描述信息,包括质量参数、分类学注释和基因组特征,已公开发布于 Figshare:https://doi.org/10.6084/m9.figshare.30920126;
“EstMB MAGdb Archaea-273” 集合中所有组装古菌基因组的 Prokka 注释结果 已公开发布于 Figshare:https://doi.org/10.6084/m9.figshare.29329166;
GitHub网址:https://github.com/Chartiza/ArchaeaDraftGenomes。
流程概述
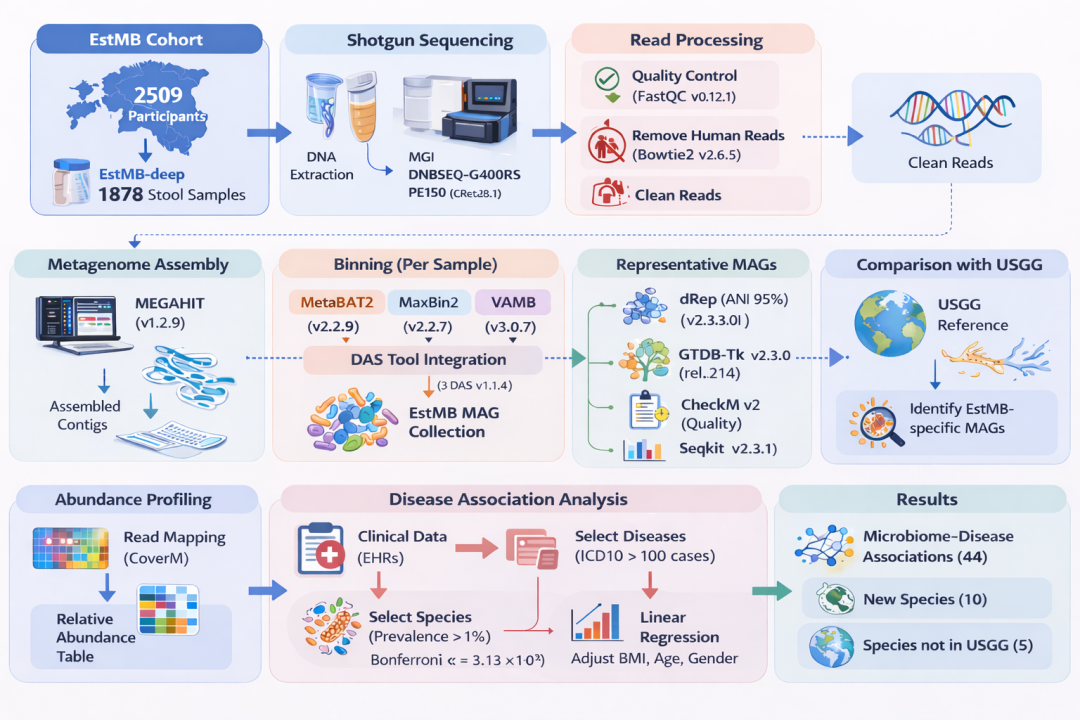
流程脚本
脚本名称 | 功能说明 |
|---|---|
Reads_preprocessing_RemoveLQ.sh | 使用 fastp 从测序数据中去除低质量 reads、 |
Reads_preprocessing_RemoveHost.sh | 使用 Bowtie2 从测序数据中去除宿主来源的 reads |
MAG_assembly.sh | 使用 MEGAHIT 从过滤后的 reads 中进行组装 |
MAG_binning.sh | 使用 MetaBAT2、MaxBin2 和 VAMB 对 contig 进行 binning,并通过 DAS Tool 进行结果整合与优化 |
MAG_clustering.sh | 使用 dRep 在物种水平对 MAG 进行去冗余聚类(ANI 阈值 >95%) |
Taxonomic_annotation.sh | 使用 GTDB-Tk v2(GTDB release 226)对 MAG 进行分类学注释 |
MAGs_quality.sh | 使用 CheckM2 评估 MAG 的质量和完整度 |
Prevalence_and_abundance.sh | 使用 CoverM 计算代表性物种在爱沙尼亚人群中的出现率和相对丰度 |
parse_CoverM_results.py | 汇总 CoverM 输出结果,生成跨样本的统计汇总表 |
Functional_annotation.sh | 使用 Prokka 对 MAG 进行功能注释 |
Paper_figures_and_stats.ipynb | 生成论文中报告的所有图表和统计结果 |
学习建议
○ 重点理解人群队列宏基因组研究的整体设计思路。 该研究利用大规模人群队列样本构建微生物参考基因组资源,并结合健康记录数据开展疾病关联分析,这种“宏基因组 + 人群健康数据”的研究框架值得重点学习;
○ 系统学习 MAG 重建的标准分析流程。 包括 reads 质控、宿主序列去除、contig 组装、binning、MAG 质量评估、物种聚类及分类学注释等关键步骤,这些流程构成当前宏基因组基因组重建研究的核心方法体系;
○ 关注人群特异性微生物参考数据库的构建策略。 文章展示了如何通过 de novo 组装发现潜在新物种并构建人群特异性参考库,对于提升宏基因组物种解析能力具有重要意义;
○ 理解宏基因组与疾病关联分析的方法。 通过将微生物丰度数据与电子健康记录结合,利用统计模型分析微生物与常见疾病之间的关联,为开展微生物组流行病学研究提供了重要参考;
○ 学习古菌在宏基因组研究中的挖掘策略。 该研究专门构建了古菌 MAG 资源集,为探索肠道古菌多样性及其潜在生物学功能提供了新的研究思路。
参考文献
Pantiukh, K., Org, E. Human gut archaea collection from Estonian population. Sci Data (2026). https://doi.org/10.1038/s41597-026-06742-1
Pantiukh K, Aasmets O, Krigul KL. et al. Metagenome-assembled genomes of Estonian Microbiome cohort reveal novel species and their links with prevalent diseases. bioRxiv(2024). doi:10.1101/2024.07.06.602324.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号