scRNA | scTCR中 T细胞动态变化(Startrac)vs scRNA指数评分

scRNA | scTCR中 T细胞动态变化(Startrac)vs scRNA指数评分

生信补给站
发布于 2024-07-12 12:30:49
发布于 2024-07-12 12:30:49
前面单细胞免疫组库VDJ|和Nature学STARTRAC,定量T细胞动态变化介绍了2018年NATRUE 文章中的STARTRAC方法,可以应用于单细胞免疫组库数据来揭示T细胞动态变化的分析。可以定量刻画T细胞的组织分布、克隆扩增情况、组织迁移和状态变化等。
scRNA分析|使用AddModuleScore 和 AUcell进行基因集打分,可视化,scRNA|使用scMetabolism完成单细胞代谢激活分数估计也介绍了使用AUCell 和 特定基因集对单细胞转录组数据进行评分。
那两者结合在一起呢?
就是SCI很常见的分析内容,结合Startrac得到的T细胞相关指数(克隆,迁移,状态变化等)与 “目标指数” 之间的相关分析内容。
一 载入R包,数据
library(tidyverse)
library(Seurat)
library(ggsci)
library(Startrac)
library(tictoc)
library(data.table)
library(ggrepel)
library(GSVA)
library(openxlsx)使用单细胞免疫组库VDJ|和Nature学STARTRAC,定量T细胞动态变化得到的Startrac输入文件以及单细胞转录组数据。或者后台回复“指数”获取示例数据。
首先对T细胞亚群进行标准的降维聚类分析
#载入数据
load("Step3.Paper_subT.Rdata")
#T 细胞 聚类分群
subT <- NormalizeData(subT)
#高变基因
subT <- FindVariableFeatures(subT, selection.method = "vst", nfeatures = 2000)
#归一化
subT <- ScaleData(subT)
#降维聚类
subT <- RunPCA(subT, npcs = 30)
subT <- FindNeighbors(subT, dims = 1:20)
subT <- FindClusters(subT, resolution = 0.5)
subT <- RunUMAP(subT, dims = 1:20)
p1 <- DimPlot(subT,group.by = "cluster_name")
p2 <- DimPlot(subT,group.by = "Clone_Num",cols = c("steelblue4","steelblue3","steelblue2","grey90"))
p1 + p2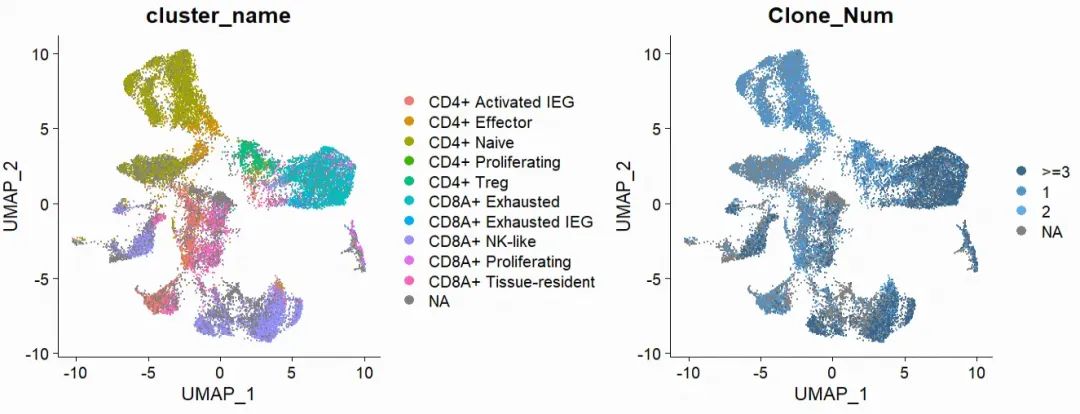
本文仅为示例,因此没有使用T细胞亚型的marker进行重新注释 ,直接使用的文献注释结果 。自己的数据需要首先进行自动注释单细胞工具箱|singleR-单细胞类型自动注释(含数据版)或者手动注释(scRNA分析|Marker gene 可视化 以及 细胞亚群注释--你是如何人工注释的?)。
二 scTCR-Startrac
可以根据单细胞免疫组库VDJ|和Nature学STARTRAC,定量T细胞动态变化得到的数据。或者后台回复“指数”获取示例数据。
load("Step2.Startrac_subT_meta.Rdata")
head(subT.meta)
#进行Startrac分析
tic("Startrac.run")
out2 <- Startrac.run(subT.meta, proj="CRC",verbose=F)
三 Startrac结果 + index score
完成Startrac分析后,结合单细胞转录组的各种评分进行分析。
1,计算细胞亚型的克隆细胞比例 与 expa的关系
也可以使用startrac的其他结果
#细胞类型的比例
subT_stat <- subT@meta.data %>%
select(cluster_name,sample,region,Clone_ID, Clone_Num,Clone_NUM)
result <- subT_stat %>%
filter(Clone_NUM >= 2) %>%
group_by(cluster_name) %>%
summarize(n_clone = n() ) %>%
mutate(percentage = n_clone / nrow(subT_stat))
dat.expa <- as.data.table(out2@cluster.sig.data)[aid==out2@proj,] %>%
filter(index == "expa")
data_in <- cbind(dat.expa,result)
ggplot(data_in,aes(x=percentage,y=value,col=majorCluster))+
geom_point(size=6) +
labs(x = "Frequency of proliferating cells" , y = "STARTRAC-expa") +
theme_classic() +
theme(legend.position = "",
panel.border = element_rect(fill=NA,color="black", size=1, linetype="solid")) +
scale_color_npg() +
geom_text_repel(aes(label = cluster_name), color = "black", vjust = -0.3, size = 5)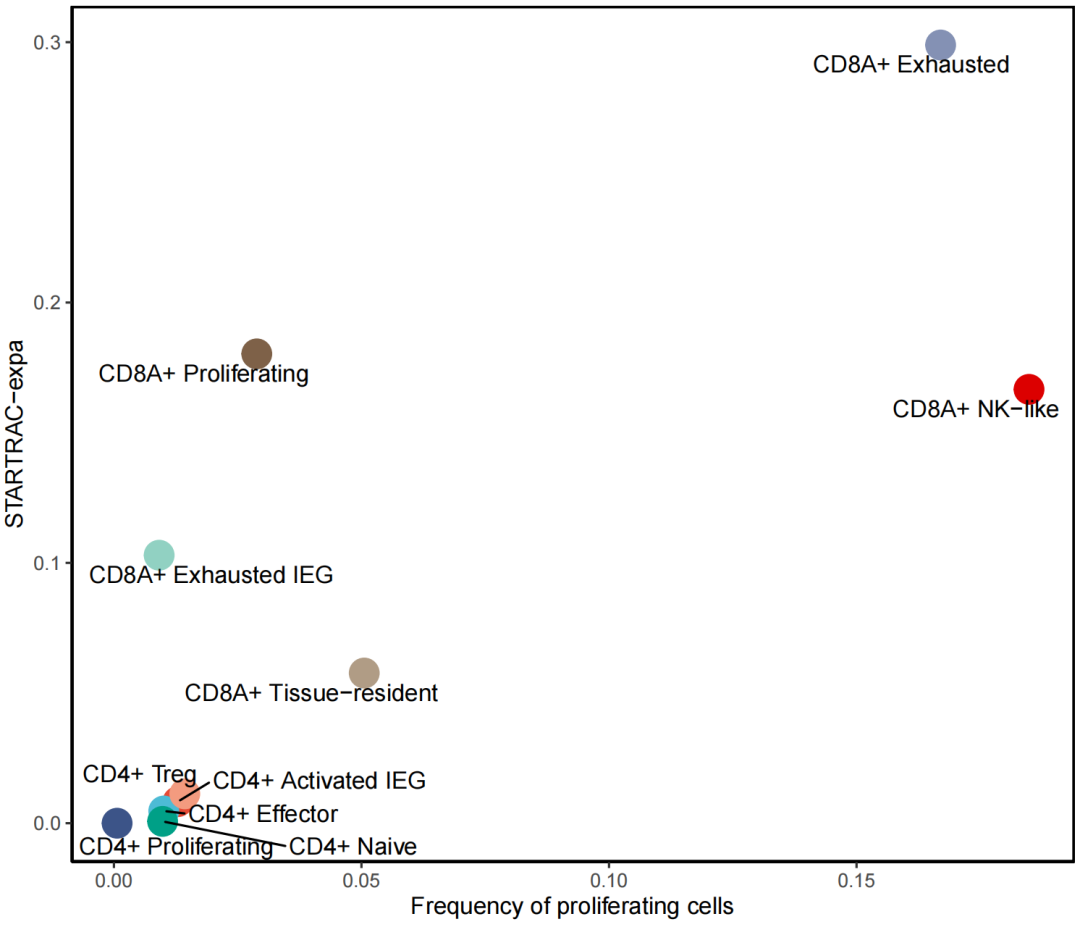
根据需要调整名字 和 配色等,比如这里左下角的只展示CD4
data_in$cluster_name[data_in$cluster_name %in% c("CD4+ Activated IEG",
"CD4+ Effector","CD4+ Naive",
"CD4+ Proliferating")] <- ""
data_in$cluster_name[data_in$cluster_name == "CD4+ Treg"] <- "CD4 T"
ggplot(data_in,aes(x=percentage,y=value,col=majorCluster))+
geom_point(size=6) +
labs(x = "Frequency of proliferating cells" , y = "STARTRAC-expa") +
theme_classic() +
theme(legend.position = "",
panel.border = element_rect(fill=NA,color="black", size=1, linetype="solid")) +
scale_color_npg() +
geom_text_repel(aes(label = cluster_name), color = "black", vjust = 0.5, size = 4)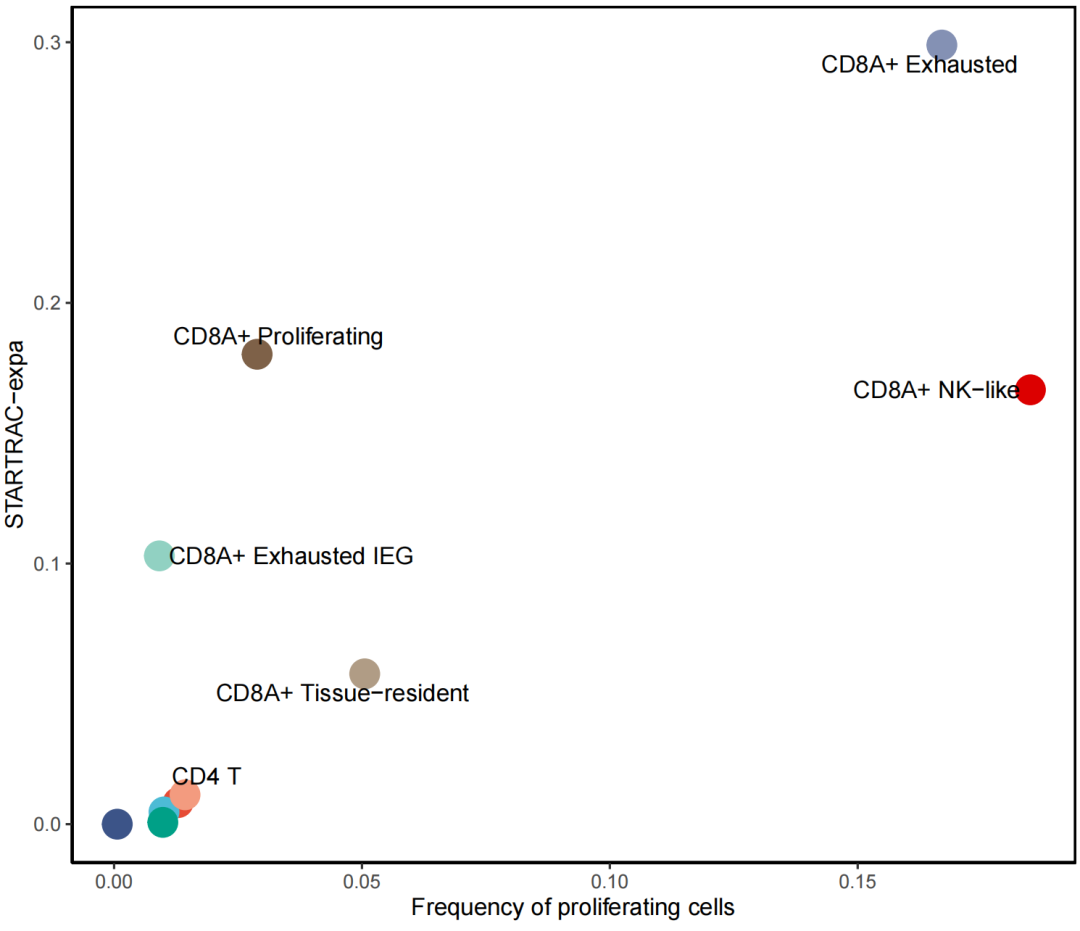
2,使用GSVA计算目标指数
将关心的基因集合(MsigDB,通路,各种分析得到的基因等)整理为列表形式,使用GSVA计算得分然后和startrac指数进行比较。
########geneList###############
geneList <- read.xlsx("geneset.xlsx",sheet = 1)
head(geneList,4)
#Pathway Genes
#RECEPTOR_INTERACTION LAMC3
#RECEPTOR_INTERACTION CHAD
#RECEPTOR_INTERACTION COL1A1
#RECEPTOR_INTERACTION COL1A2
geneset = split(geneList$Genes,geneList$Pathway) #转为list
#进行gsva分析
#指定细胞类型的列为Idents ,然后计算均值
Idents(subT) <- "cluster_name"
#计算celltype均值
cluster.averages <- AverageExpression(subT)
#注意表达谱exp载入后需转化为matrix,前面已转换
exp <- as.matrix(cluster.averages$RNA )
re <- gsva(exp, geneset, method="ssgsea",
mx.diff=FALSE, verbose=FALSE)
#合并scTCR数据
data_in2 <- re %>%
t() %>%
as.data.frame() %>%
rownames_to_column("majorCluster") %>%
inner_join(dat.expa)
ggplot(data_in2,aes(x=RECEPTOR_INTERACTION,y=value,col=majorCluster))+
geom_point(size=6) +
labs( y = "STARTRAC-expa") +
theme_classic() +
theme(legend.position = "",
panel.border = element_rect(fill=NA,color="black", size=1, linetype="solid")) +
scale_color_npg() +
geom_text_repel(aes(label = majorCluster), color = "black", vjust = -0.3, size = 4)
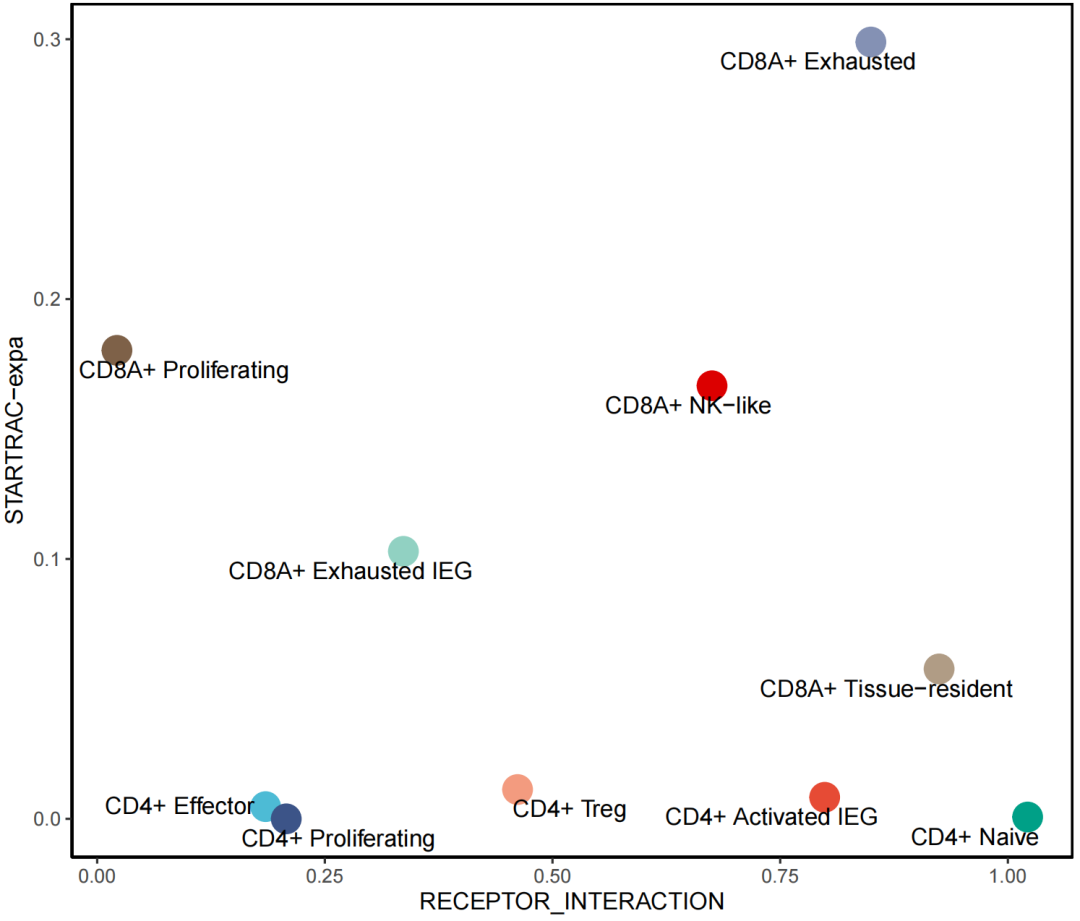
以上就完成了目标指数与T细胞动态相关的分析。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-07-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号