富集分析-KEGG/GO
原创

·KEGG(Kyoto Encyclopedia of Genes and Genomes)
是系统分析基因功能、基因组 信息数据库,它有助于研究者把基因及表达信息作为一个整体 网络进行研究,以“理解生物系统的高级功能和实用程序资源库”著称。
·基因本体(Gene Ontology,GO)
是一个在生物信息学领域中广泛使用的本体。1998年由研究三种模式(果蝇、小鼠和酵母)基因组的研究者共同发起组织了一个称为基因本体联盟的专业团队。创建基因本体的初衷是希望提供一个可具代表性的规范化的基因和基因产物特性的术语描绘或词义解释的工作平 台 。现在已包含数十个动物、植物、 微生物的数据库。基因本体涉及的基因和基因产物词汇分为三大 类,涵盖生物学的三个方面:
• 细胞组分(cellular component)(cc): 细胞的每个部分和细胞外环境。
• 分子功能(molecular function)(mf): 可以描述为分子水平的活性,如催化或结合活性。
• 生物过程(biological process)(bp): 生物过程系指由一个或多个分子功能有序组合而产生的系列事 件。其定义有广义和狭义之分,在词义上可以区分为泛指和特指。一般规律 是,一个过程是由多个不 同的步骤组成。
·通过将差异基因做 GO 富集分析,可以把基因按照不同的功能进行归类,达到对基因进行注释和分类的目的。
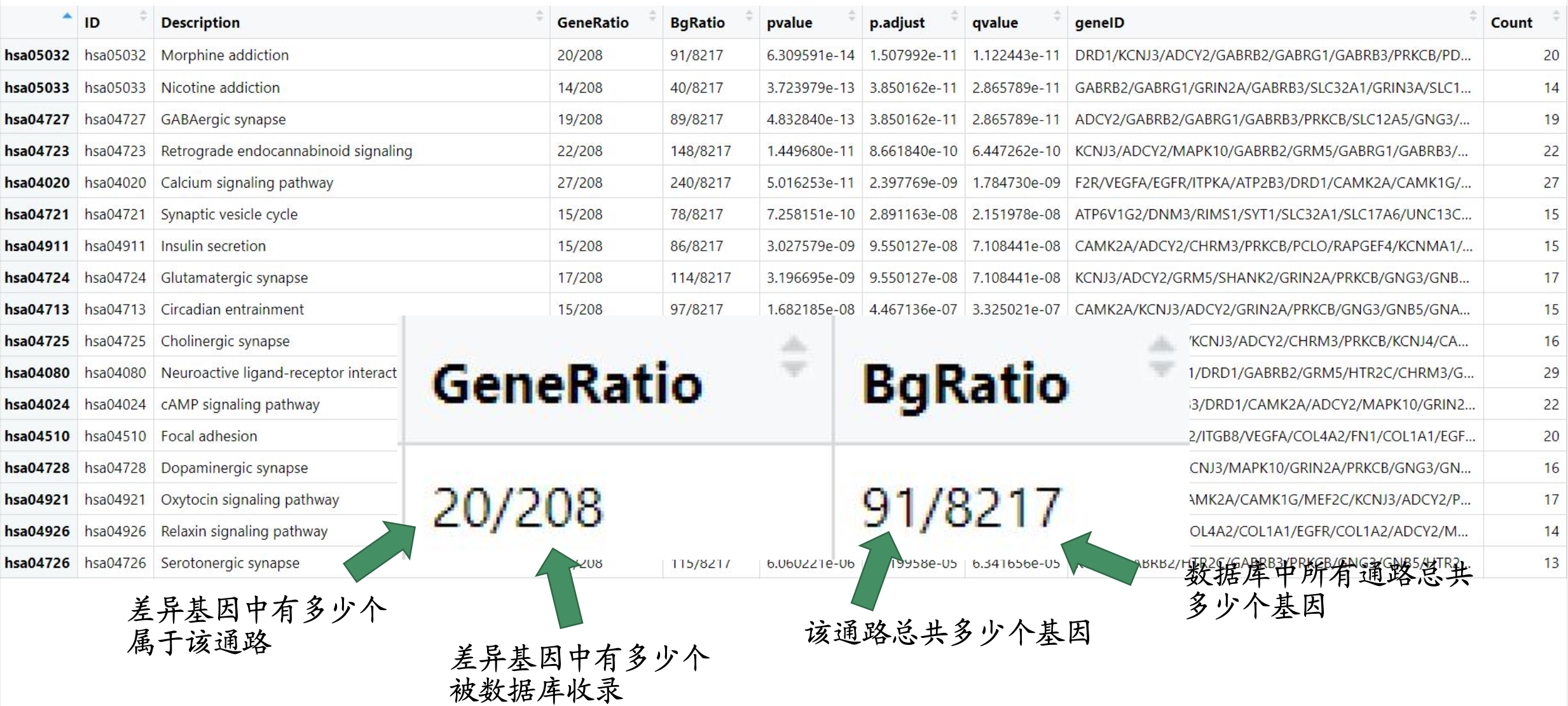
富集分析数据格式
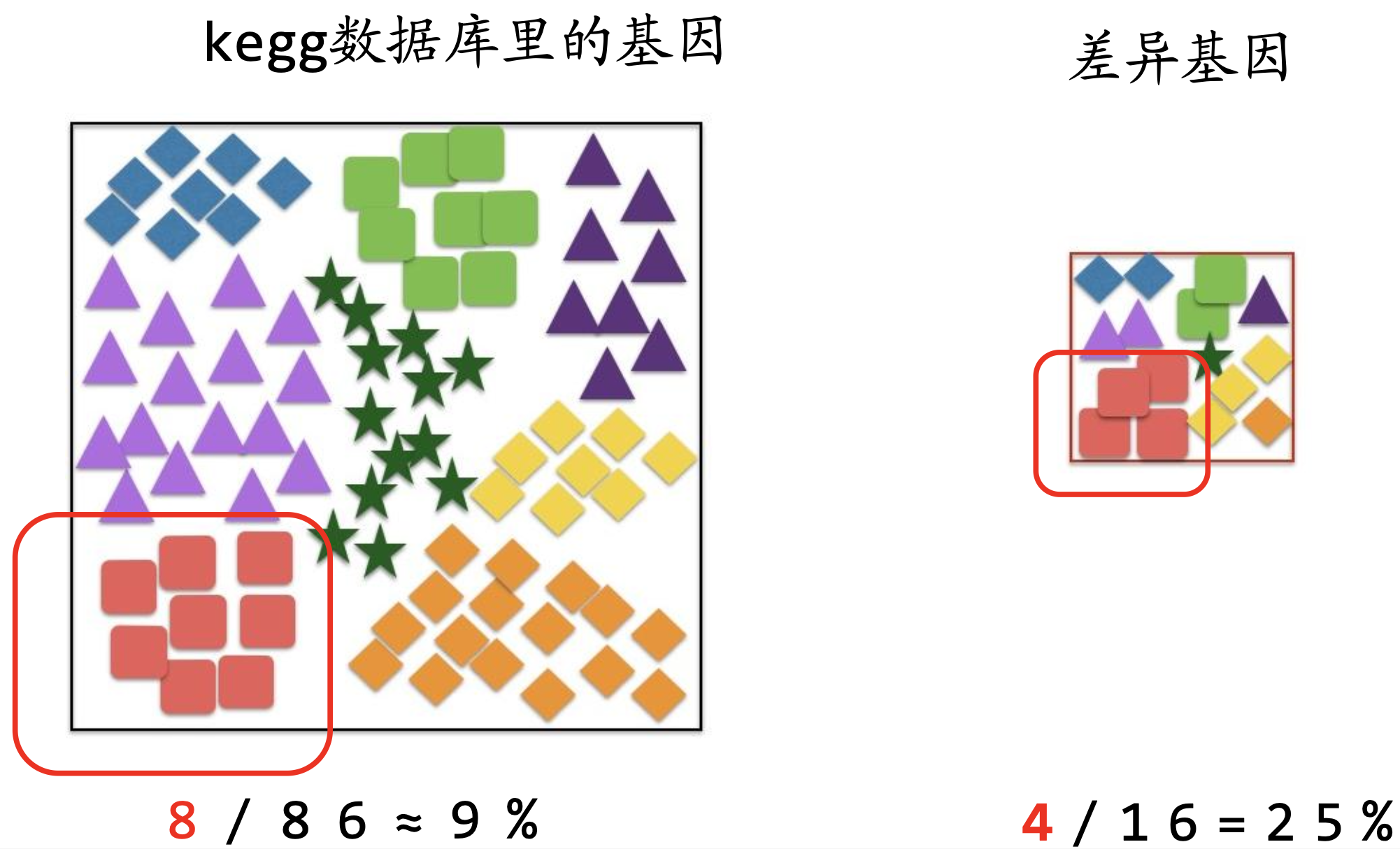
富集分析的理解
富集分析-衡量每个通路里的基因在差异基因里是否足够多
富集分析代码
rm(list = ls())
load(file = 'step4output.Rdata')
library(clusterProfiler)
library(ggthemes)
library(org.Hs.eg.db)
library(dplyr)
library(ggplot2)
library(stringr)
library(enrichplot)1.GO 富集分析----
#(1)输入数据 组织输入数据
gene_up = deg$ENTREZID[deg$change == 'up']
gene_down = deg$ENTREZID[deg$change == 'down']
gene_diff = c(gene_up,gene_down)#(2)富集
#以下步骤耗时很长,设置了存在即跳过
f = paste0(gse_number,"_GO.Rdata") #如果存在文件ff,就会跳过,直接load(f)
if(!file.exists(f)){
ego <- enrichGO(gene = gene_diff,
OrgDb= org.Hs.eg.db, #转换ID的数据库
ont = "ALL", #取子集
readable = TRUE) #可以直接讲结果转化为simple
ego_BP <- enrichGO(gene = gene_diff,
OrgDb= org.Hs.eg.db,
ont = "BP", #单独做某一个分支,取子集
readable = TRUE)
#ont参数:One of "BP", "MF", and "CC" subontologies, or "ALL" for all three.
save(ego,ego_BP,file = f)
}
load(f)#(3)可视化
#条带图
barplot(ego) #直接拿富集结果来画图
barplot(ego, split = "ONTOLOGY", font.size = 10,
showCategory = 5) +
facet_grid(ONTOLOGY ~ ., space = "free_y",scales = "free_y") #结果看图a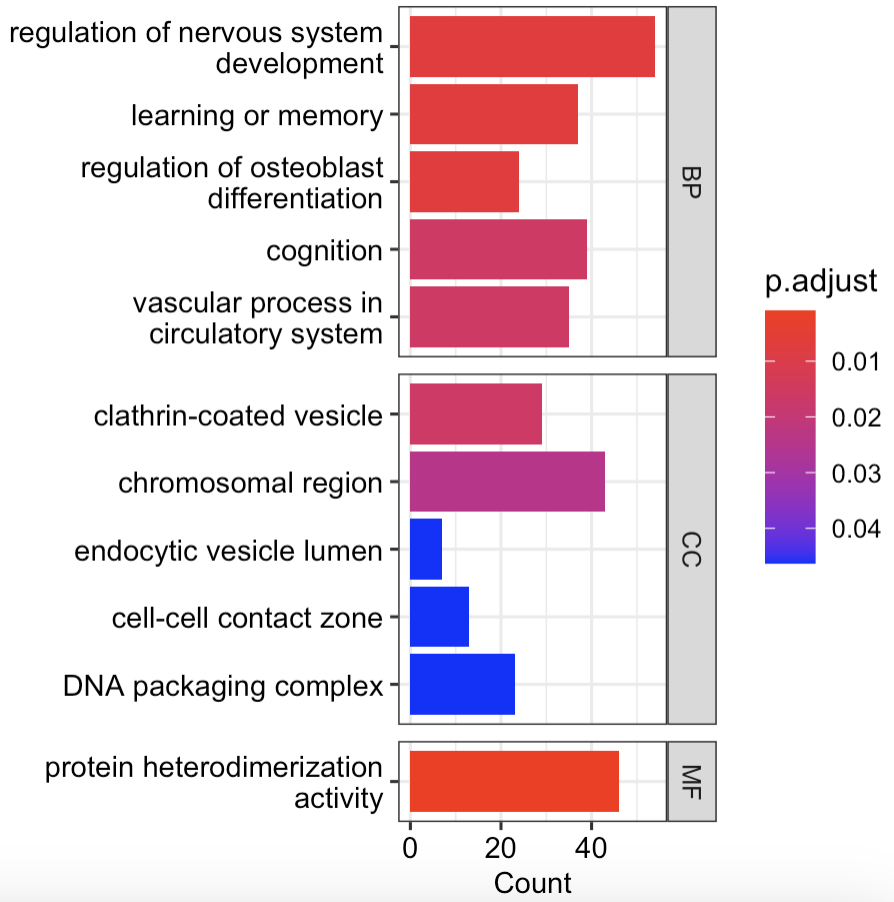
图a
气泡图
dotplot(ego)
dotplot(ego, split = "ONTOLOGY", font.size = 10,
showCategory = 5) +
facet_grid(ONTOLOGY ~ ., space = "free_y",scales = "free_y") 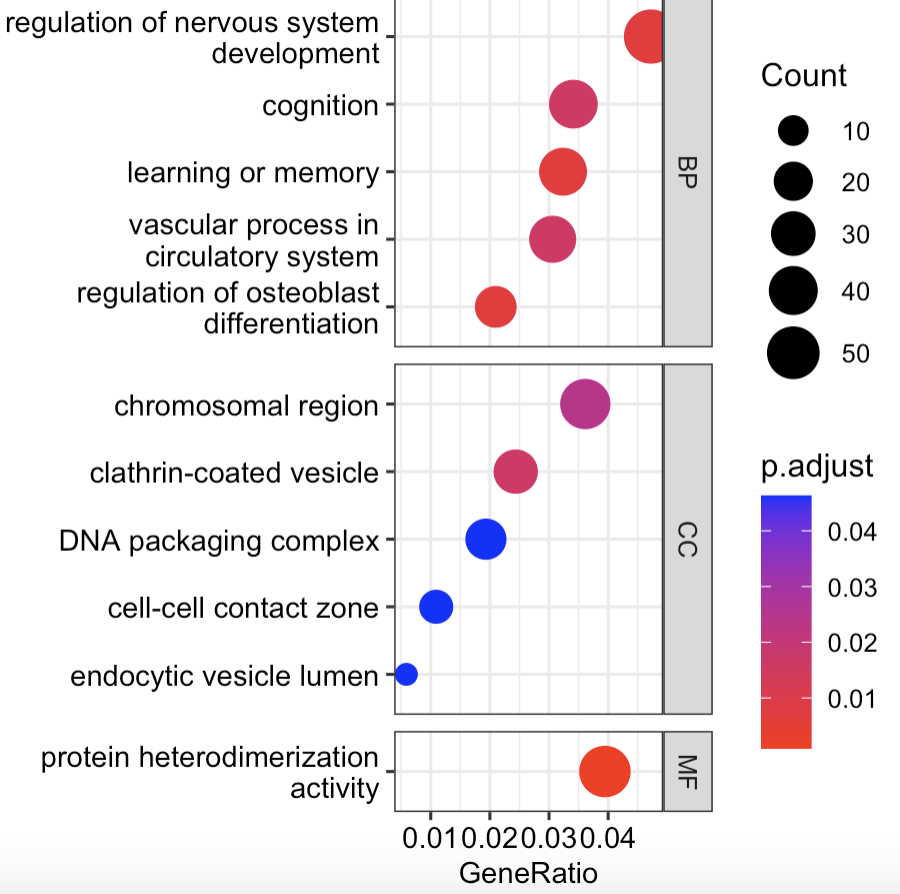
气泡图
(3)展示top通路的共同基因,要放大看。
#gl 用于设置下图的颜色
gl = deg$logFC
names(gl)=deg$ENTREZID
#Gene-Concept Network,要放大看
cnetplot(ego,
#layout = "star", #用layout()就换了一种视图圆形的(图f)
color.params = list(foldChange = gl),
showCategory = 3)
#结果图egoplot(ego_BP) # 用于展示通路-通路之间的联系(图g)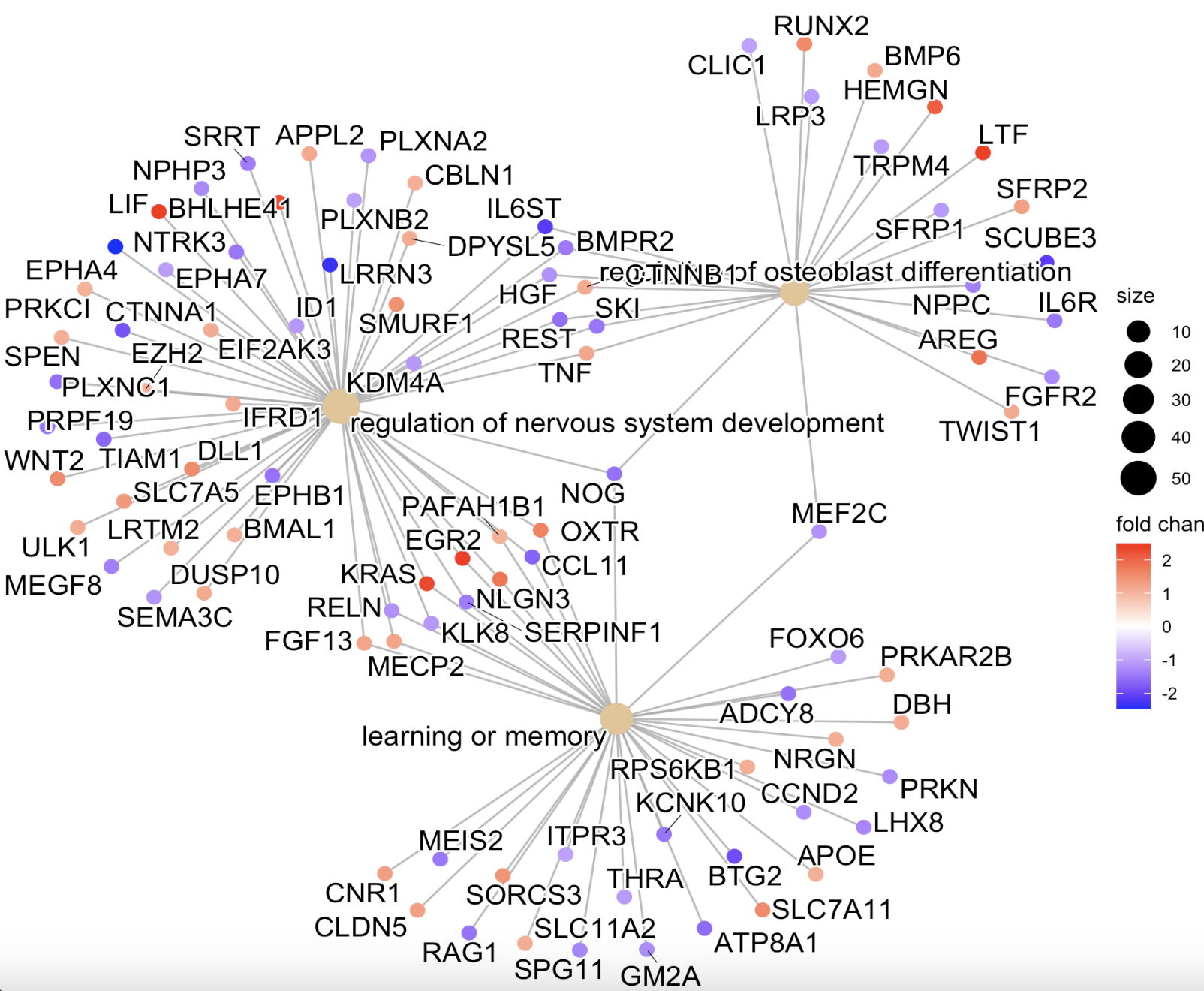
图e
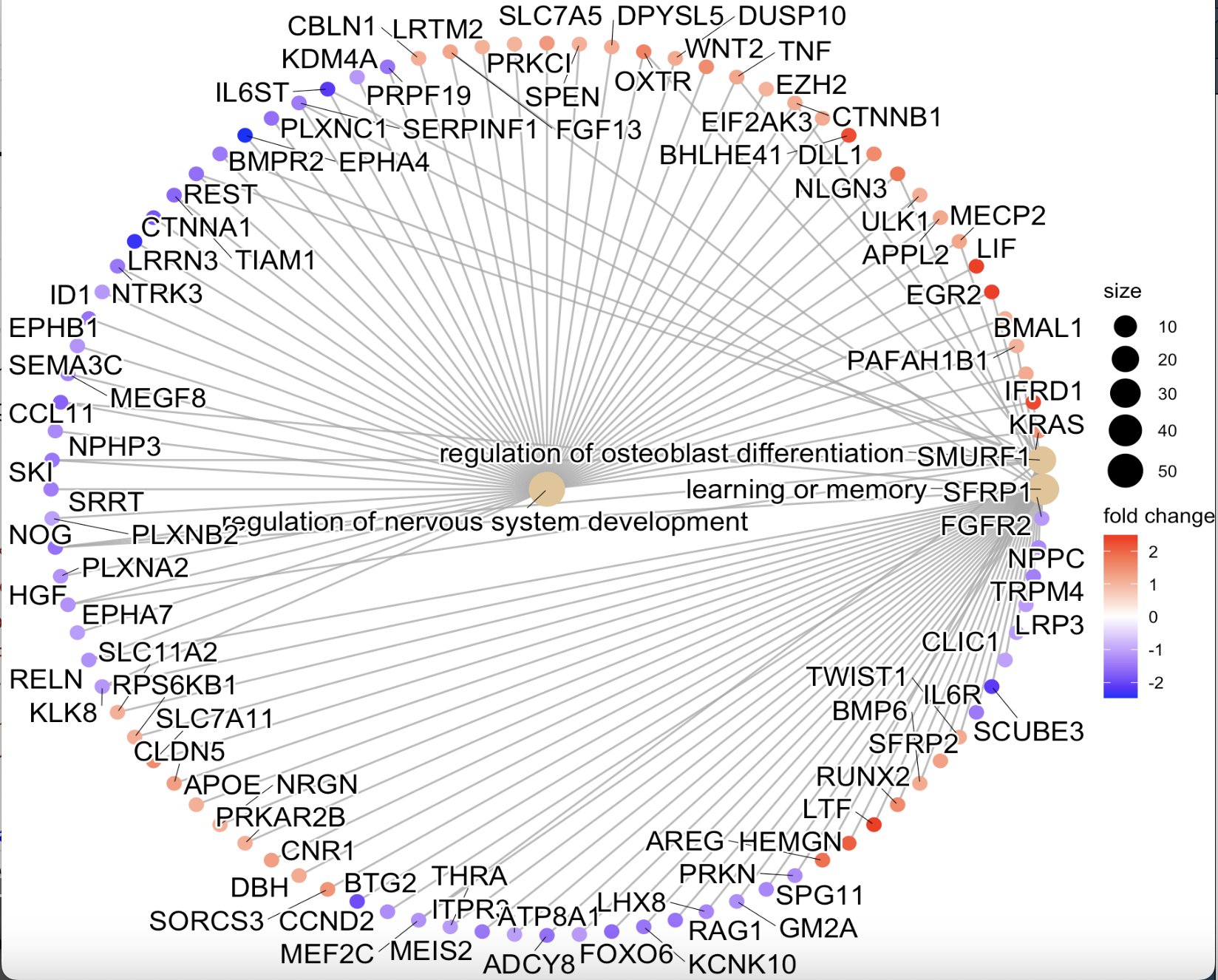
图f
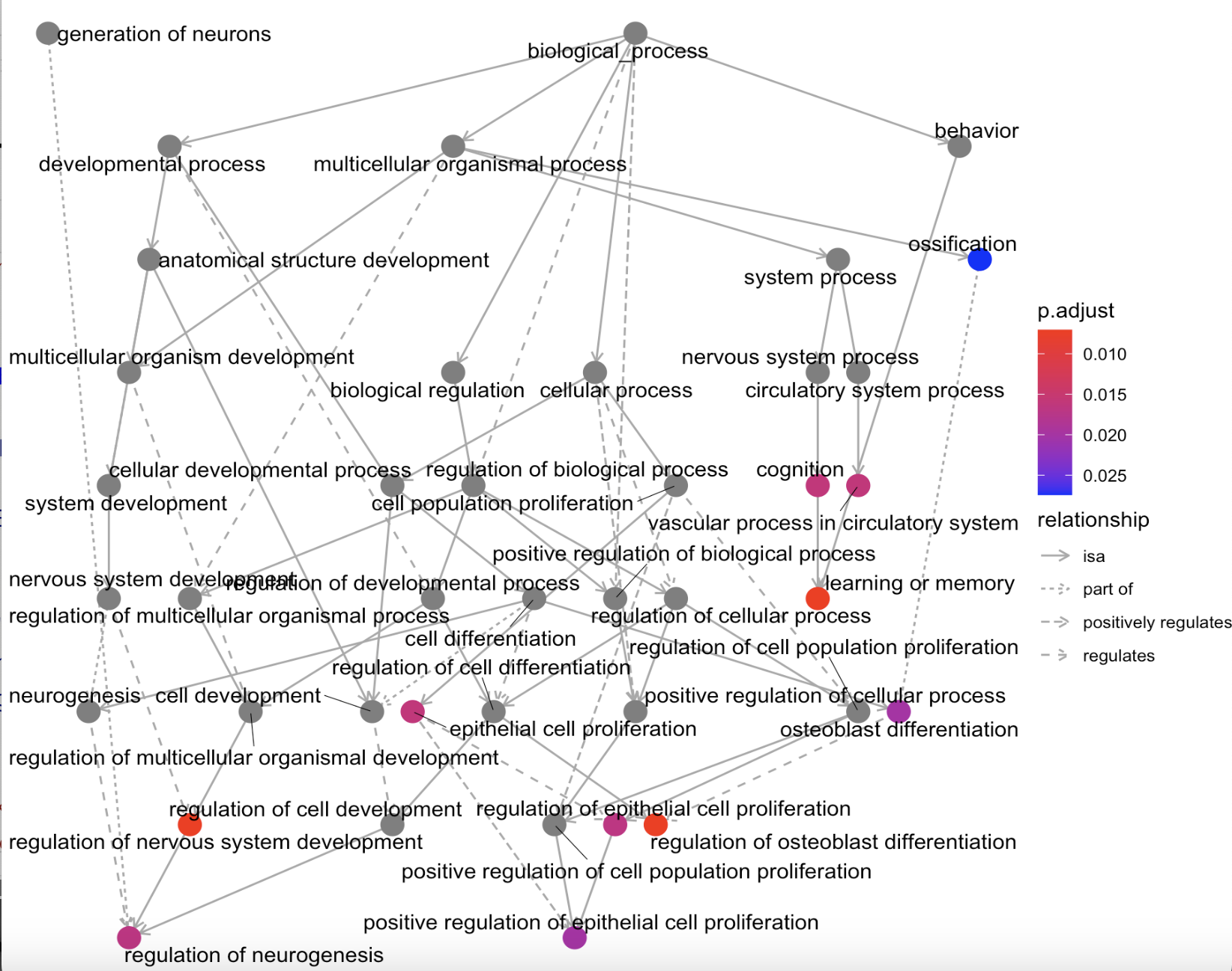
图g
2.KEGG pathway analysis----
#上调、下调、差异、所有基因
#(1)输入数据 与GO不需要作区分是完全一样的
gene_up = deg[deg$change == 'up','ENTREZID']
gene_down = deg[deg$change == 'down','ENTREZID']
gene_diff = c(gene_up,gene_down)
#(2)对上调/下调/所有差异基因进行富集分析 (包含存在即跳过if...)
f2 = paste0(gse_number,"_KEGG.Rdata")
if(!file.exists(f2)){
kk.up <- enrichKEGG(gene = gene_up, #富集-enrichKEGG()
organism = 'hsa') #指定物种的名称,人类-hsa,Go中是ORGDB
kk.down <- enrichKEGG(gene = gene_down,
organism = 'hsa')
kk.diff <- enrichKEGG(gene = gene_diff,
organism = 'hsa') #一共做了三次富集
save(kk.diff,kk.down,kk.up,file = f2)
}
load(f2)
#KEGG 经常存在一个问题就是没有富集到,所以要看看富集到了没
#(3)看看富集到了吗?https://mp.weixin.qq.com/s/NglawJgVgrMJ0QfD-YRBQg
> table(kk.diff@result$p.adjust<0.05)
FALSE TRUE
320 2
> table(kk.up@result$p.adjust<0.05)
FALSE TRUE
292 3
> table(kk.down@result$p.adjust<0.05)
FALSE
290
#没有富集到 不满足p.adjust值,是正常的(这是Y叔的包,只能用p.adjust富集,但有的满足p值,可以通过p值挽救一下。或者换一种方式富集,或者调整差异基因的阈值后面会有)
# 富集分析所有图表默认都是用p.adjust,富集不到可以退而求其次用p值,在文中说明即可(4)双向图 上下调基因分开画图
source("kegg_plot_function.R") #source 是在不打开脚本的情况下运行,也是画图函数,是自定义函数(自己写的)
g_kegg <- kegg_plot(kk.up,kk.down) #可以根据不同阈值画图看图h,默认p值为0.05
g_kegg
#g_kegg +scale_y_continuous(labels = c(2,0,2,4,6)) #强行修改横坐标上的标签,删掉#可把图h中的-2改成2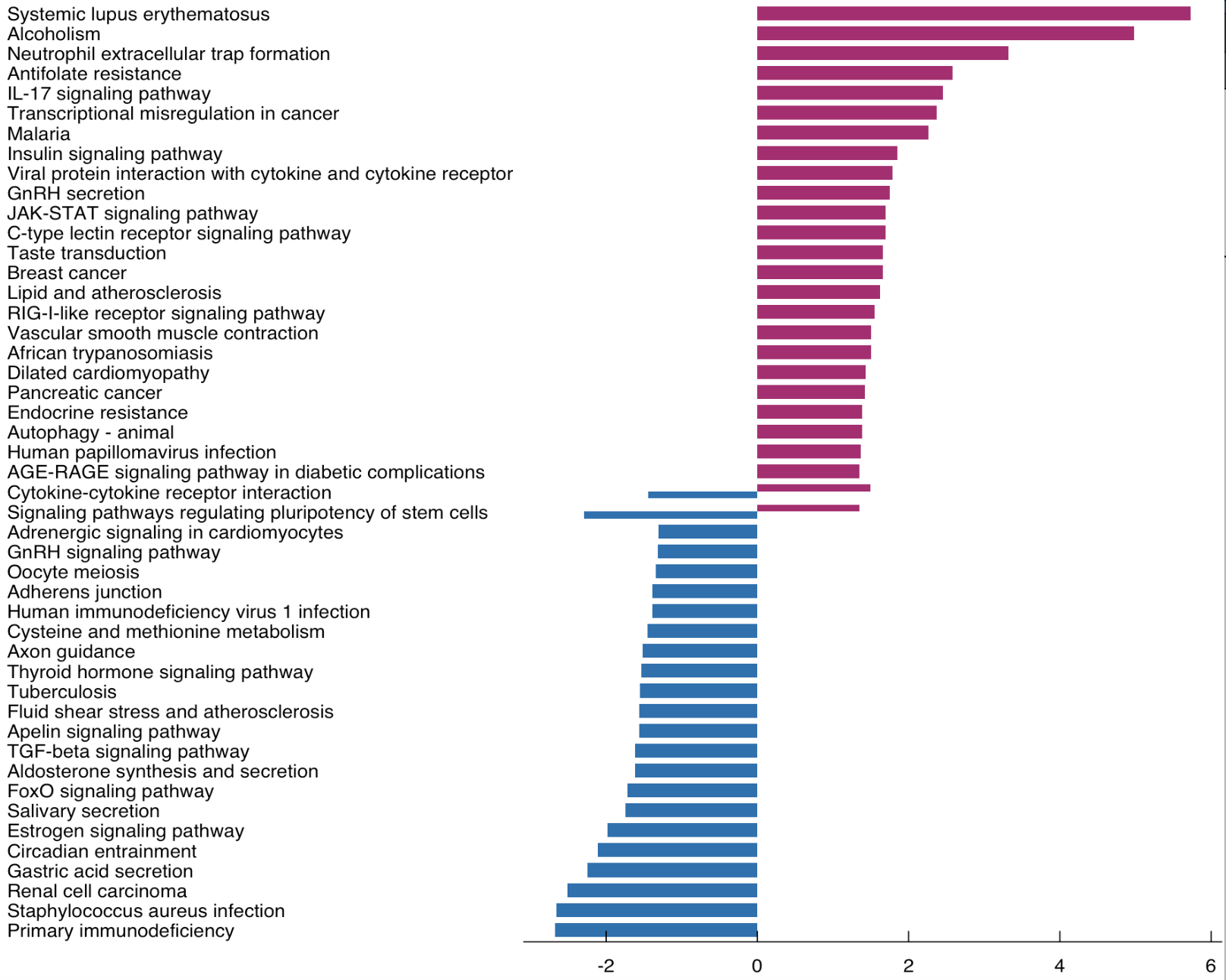
双向图
图h
3.能看懂的资料越来越多----
GSEA:https://www.yuque.com/docs/share/a67a180f-dd2b-4f6f-96c2-68a4b86fe862?
富集分析学习更多:http://yulab-smu.top/clusterProfiler-book/index.html
弦图:https://www.yuque.com/xiaojiewanglezenmofenshen/dbwkg1/dgs65p
GOplot:https://mp.weixin.qq.com/s/LonwdDhDn8iFUfxqSJ2Wew
问题数据和常见错误分析
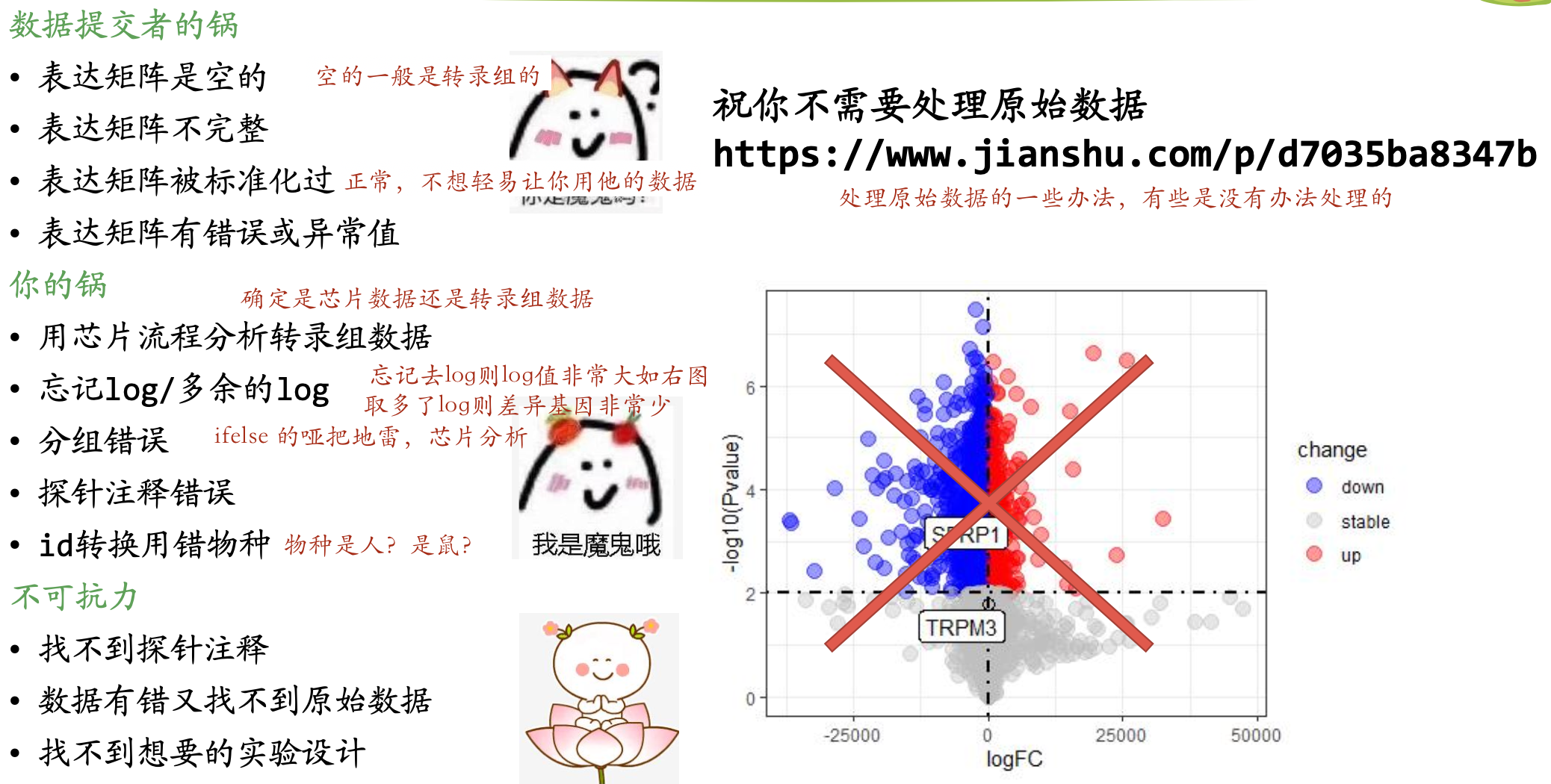
问题数据和常见的错误分析
复杂的数据类型
1多分组数据
·一个对照,2+ 实验组
·两两差异分析
·需要做的事情:向量取子集;矩阵取子集
2多数据联合分析
·两种选择做:
·1分别分析:各自差异分析,差异基因取交集
·2先合并,后差异分析:
·原则上选择来自同一个芯片平台的GSEGSE
·需要处理批次效应,BatchBatch effectt 图n
·不要选择一个全是处理组,一个全是对照组的数据去合并
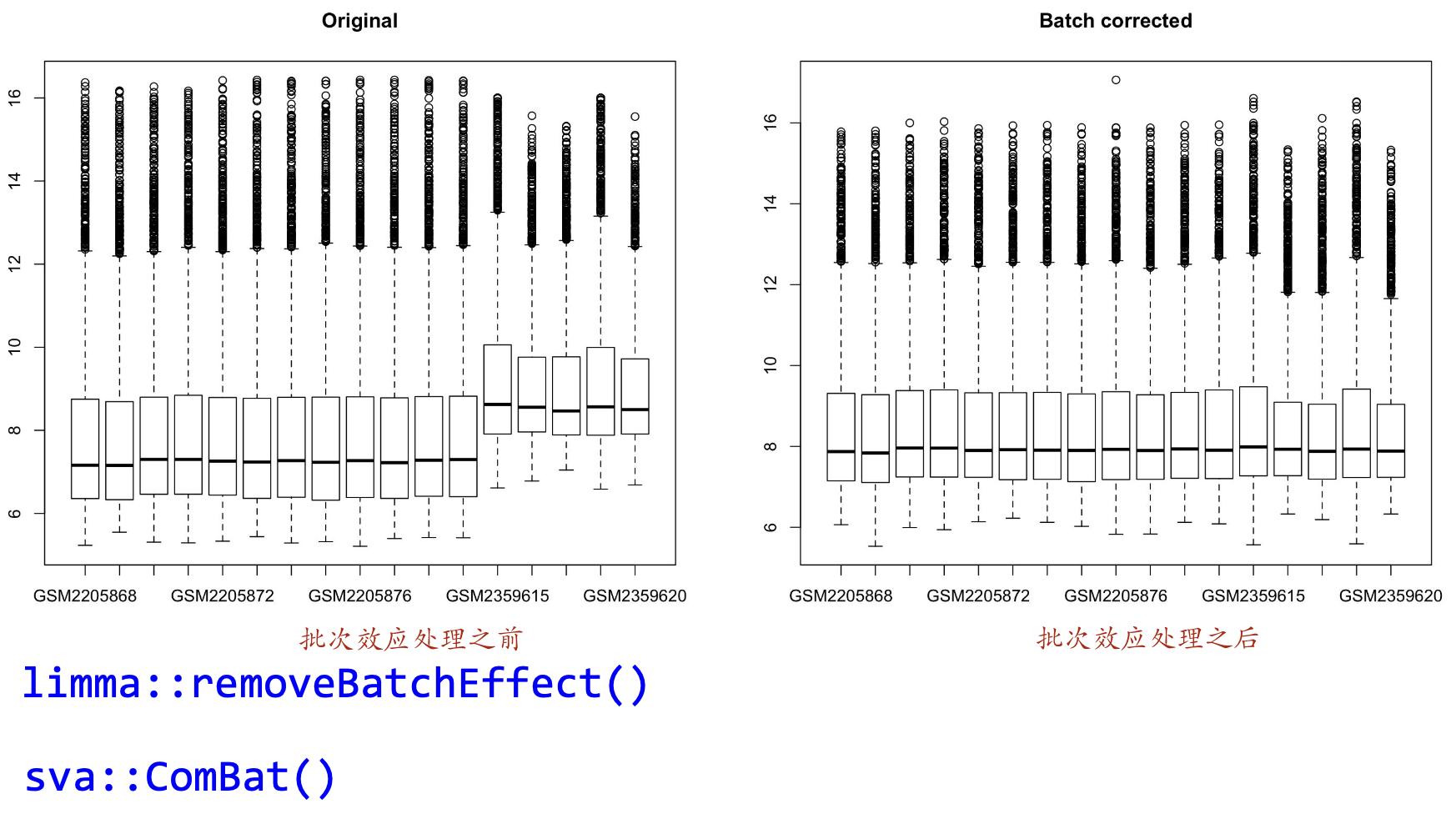
图n
两个处理批次效应的包
illma::removeBatchEffect()
sva::ComBat()#标准流程的后续
PPI网络 蛋白蛋白相互作用的网络-网页工具string
输入数据:差异基因
输出数据:一个ppi图,可以导出数据(从ppi中得到数据)
将得到的数据放入cytoscape中,进行网络可视化
之后还可以寻找hub基因(核心基因),插件cytohHubba 不涉及R语言
子网络从大网络中拆分小网络 插件Mcode
制作string输入数据的代码
-----来自生信技能树----
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
作者已关闭评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号