【生信分析】免疫组库基础分析8-CDR3氨基酸使用频率

【生信分析】免疫组库基础分析8-CDR3氨基酸使用频率

三兔测序学社
发布于 2025-10-20 17:02:13
发布于 2025-10-20 17:02:13
1.CDR3氨基酸使用频率计算
CDR3区域的20种氨基酸使用频率计算,首先需要知道20种氨基酸种类,有两种计算方法。
第一种方法:不考虑每一个序列的相对频率差异,每一条序列都一样。先算出每一条序列CDR3中这20种氨基酸出现的次数,每一种氨基酸进行累积后,再进行20种氨基酸的相对频率计算。
第二种方法:加权计算:考虑每一个序列的相对频率差异。将每一条序列CDR3中这20种氨基酸出现的次数乘以该序列的相对频率,得到加权次数。然后每一种,每一种氨基酸进行累积后,再进行20种氨基酸的相对频率计算,得到加权频率结果。
代码举例说明上述分析思路
# 加载必要的可视化包
library(ggplot2)
# 定义标准20种氨基酸(生物化学标准)
standard_aas <- c("G", "A", "S", "C", "D", "P", "N", "T", "E", "V",
"Q", "H", "M", "I", "L", "K", "R", "F", "Y", "W")
# 创建包含10个虚拟CDR3序列及其频率的数据框
# 序列长度在8-20个氨基酸之间,频率总和为100%
seq_data <- data.frame(
Sequence = c(
"CASSLGNTGQLY-F", # 序列1:长度13
"CASSQETGQ*YF", # 序列2:长度11
"CASSEHGQLYF", # 序列3:长度11
"CASSLkQGNTIYF", # 序列4:长度13
"CASSQPGGQETQYF", # 序列5:长度14
"CASSLDVNTGQLYF", # 序列6:长度13
"CASSLEGANTGQFY", # 序列7:长度13
"CASSLGQWGNTIYF", # 序列8:长度14
"CASSQGGNKEQYF", # 序列9:长度12
"CASSLGMGQGRNTIYF" # 序列10:长度15
),
Frequency = c(15, 12, 10, 8, 20, 5, 10, 8, 7, 5) # 各序列占比(%)
)
# 验证频率总和是否为100%
if (sum(seq_data$Frequency) != 100) {
stop("错误:序列频率总和必须等于100%")
}
# [3] 核心计算函数(分步实现)
calculate_aa_stats <- function(seq_df, aas) {
# 初始化结果数据框(包含所有统计维度)
result <- data.frame(
AminoAcid = aas,
RawCount = 0, # 原始出现次数
RawFrequency = 0, # 原始频率(不考虑克隆权重)
WeightedCount = 0, # 加权计数(考虑克隆频率)
WeightedFrequency = 0, # 最终加权频率
stringsAsFactors = FALSE
)
# [3.1] 原始计数计算
total_aa <- 0 # 总氨基酸计数
for(seq in seq_df$Sequence) {
chars <- strsplit(seq, "")[[1]] # 拆分为单字符
counts <- table(factor(chars, levels=aas)) # 强制统计所有氨基酸
result$RawCount <- result$RawCount + as.numeric(counts)
total_aa <- total_aa + sum(counts) # 累计总氨基酸数
}
# 计算原始频率(百分比)
result$RawFrequency <- round(result$RawCount / total_aa * 100, 2)
# [3.2] 加权频率计算
total_weighted <- 0 # 加权总和
for(i in 1:nrow(seq_df)) {
seq <- seq_df$Sequence[i]
weight <- seq_df$Frequency[i] / 100 # 转换为权重系数
chars <- strsplit(seq, "")[[1]]
counts <- table(factor(chars, levels=aas))
weighted_counts <- as.numeric(counts) * weight # 当前序列的加权贡献
result$WeightedCount <- result$WeightedCount + weighted_counts
total_weighted <- total_weighted + sum(weighted_counts)
}
# 计算加权频率(百分比)
result$WeightedFrequency <- round(result$WeightedCount / total_weighted * 100, 2)
return(result)
}
aa_stats <- calculate_aa_stats(seq_data, standard_aas) # 计算统计量
# [5] 结果输出(按加权频率降序)
final_result <- aa_stats[order(-aa_stats$WeightedFrequency),
c("AminoAcid", "RawFrequency", "WeightedFrequency")]
print(final_result)
# [6] 专业可视化
ggplot(final_result, aes(x=reorder(AminoAcid, -WeightedFrequency))) +
geom_col(aes(y=RawFrequency, fill="原始频率"),
width=0.35, position=position_nudge(x=-0.18)) +
geom_col(aes(y=WeightedFrequency, fill="加权频率"),
width=0.35, position=position_nudge(x=0.18)) +
geom_text(aes(y=RawFrequency, label=sprintf("%.1f%%", RawFrequency)),
vjust=-0.5, size=3, nudge_x=-0.18) +
geom_text(aes(y=WeightedFrequency, label=sprintf("%.1f%%", WeightedFrequency)),
vjust=-0.5, size=3, nudge_x=0.18) +
scale_fill_manual(values=c("#4E79A7", "#F28E2B")) +
labs(title="CDR3区域氨基酸使用频率对比",
subtitle="原始频率 vs 克隆加权频率",
x="氨基酸", y="频率(%)", fill="统计方法") +
theme_minimal(base_size=12) +
theme(
axis.text.x=element_text(angle=45, hjust=1, face="bold"),
legend.position="top",
panel.grid.major.x=element_blank()
)
输出结果如下:当克隆数量越多的时候,每一个克隆的相对频率就越小,就会导致加权频率与不加权频率(原始频率)之间似乎差异不大,但在实际过程种,由于特定克隆的扩增,理应考虑频率改变带来的影响,因此实际分析种优先选择加权频率这种计算方法。这种方法也将在后续CDR3氨基酸motif应用到。

图1:比较加权频率与原始频率(不加权)的差异
2.文献中氨基酸使用频率使用分析图
2.1 柱状图
柱状图是CDR3氨基酸使用最常见的可视化图,排列顺序通常可以根据氨基酸理化性质进行排列。下图2用疏水性大小进行了排列。
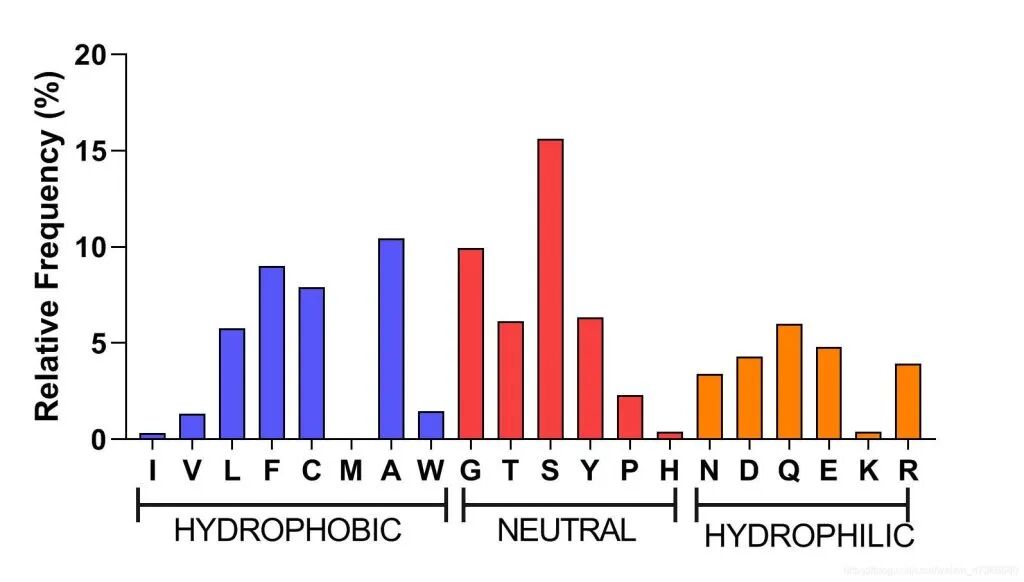
图2:CDR3氨基酸使用频率(顺序按照疏水性进行排列)
2.2 叠加柱状图(多个样本比较)
这种叠加柱状图好处:多样本比较,如下图就是20个样本进行比较,可以比较每一个样本之间的差异,找出无显著差异的趋势(可能是某些单个样本的异常导致的)。
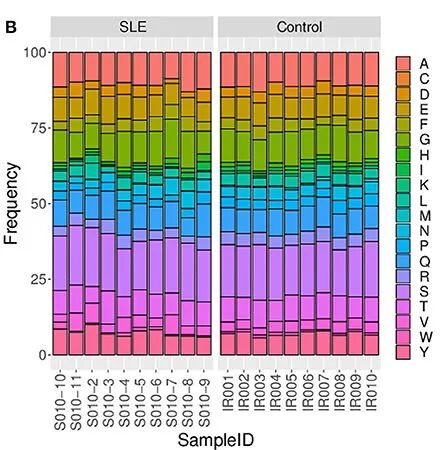
图3:SLE与对照组之间不同样本之间CDR氨基酸使用频率的比较
高级分析
如下图所示,对每一个TCR、BCR受体不同位置进行氨基酸组成分析,比较不同位置下不同氨基酸的使用差异与优势使用氨基酸种类,其实这种分析相当于CDR3氨基酸的motif分析,分析保守氨基酸(这个位点高频使用的氨基酸)及差异氨基酸(高可变区域差异氨基酸多样性比较)
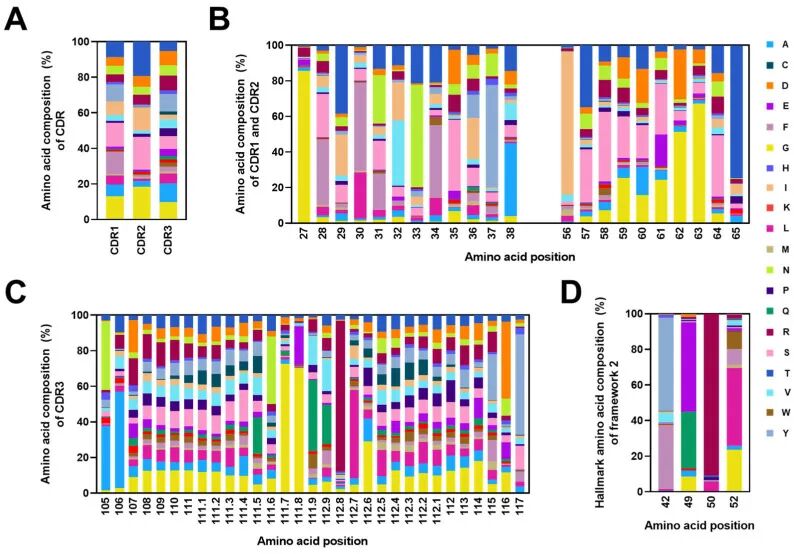
图4:VHH的CDR和FR2的氨基酸组成。(A)对CDR1、CDR2和CDR3完整序列的氨基酸组成进行综合分析。(B,C)深入研究CDR1, CDR2, CDR3中每个位置的氨基酸组成。(D) FR2中42、49、50和52位特征氨基酸组成的详细分析。在所有图(A-D)中,CDR和FR2中的氨基酸组成以百分比表示,并以不同颜色的堆叠条形显示。每个堆叠的条形图代表每个氨基酸在特定位置的比例。x轴表示氨基酸的位置与IMGT的编号一致。
图片来源:
图1-2:本文作者版权
图3来自文献:Ye X, Wang Z, Ye Q, Zhang J, Huang P, Song J, Li Y, Zhang H, Song F, Xuan Z, Wang K. High-Throughput Sequencing-Based Analysis of T Cell Repertoire in Lupus Nephritis. Front Immunol. 2020 Aug 6;11:1618. doi: 10.3389/fimmu.2020.01618. PMID: 32849548; PMCID: PMC7423971.
图4来自文献:Lee HE, Cho AH, Hwang JH, Kim JW, Yang HR, Ryu T, Jung Y, Lee S. Development, High-Throughput Profiling, and Biopanning of a Large Phage Display Single-Domain Antibody Library. Int J Mol Sci. 2024 Apr 27;25(9):4791. doi: 10.3390/ijms25094791. PMID: 38732011; PMCID: PMC11083953.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-08-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号