空间转录组: 反卷积及可视化


引言
本系列讲解 空间转录组学 (Spatial Transcriptomics) 相关基础知识与数据分析教程[1],持续更新,欢迎关注,转发,文末有交流群!
RCTD
接下来,我们使用 spacexr(也称为 RCTD)进行反卷积。默认情况下,runRctd() 的 rctd_mode = "doublet" 指定最多两个亚群共存于一个数据单元(即一个 spot)中;在此,我们将 rctd_mode 设置为 "full",以允许拟合任意数量的亚群。
rctd_data <- createRctd(vis, sce, cell_type_col = "Annogrp")
(res <- runRctd(rctd_data, max_cores = 4, rctd_mode = "full"))
## class: SpatialExperiment
## dim: 12 4962
## metadata(4): spatial_rna config cell_type_info internal_vars
## assays(1): weights
## rownames(12): B DCIS1 ... stromal tumor
## rowData names(0):
## colnames(4962): AACACCTACTATCGAA-1 AACACGTGCATCGCAC-1 ...
## TGTTGGCCAGACCTAC-1 TGTTGGCCTACACGTG-1
## colData names(1): sample_id
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## spatialCoords names(2) : x y
## imgData names(0):
由 RCTD 推断的权重应进行归一化,使得每个 spot 的细胞类型比例之和为 1:
# scale weights such that they sum to 1
ws <- assay(res)
ws <- sweep(ws, 2, colSums(ws), `/`)
ws_rctd <- data.frame(t(as.matrix(ws)))
ws_rctd[1:5, 1:5] |> round(2)
## B DCIS1 DCIS2 T dendritic
## AACACCTACTATCGAA-1 0.00 0.00 0.00 0.00 0.01
## AACACGTGCATCGCAC-1 0.04 0.01 0.00 0.04 0.00
## AACACTTGGCAAGGAA-1 0.00 0.00 0.03 0.01 0.01
## AACAGGAAGAGCATAG-1 0.03 0.00 0.00 0.08 0.03
## AACAGGATTCATAGTT-1 0.00 0.00 0.01 0.00 0.00
# add proportion estimates to colData
colData(vis)[names(ws_rctd)] <- ws_rctd[colnames(vis), ]
CARD
另一种可用的方法是 CARD。首先,我们重命名 CARD 的空间坐标列。
# realize the delayed matrices, as CARD do not support delayed matrix handling yet
counts(sce) <- as(counts(sce), "sparseMatrix")
counts(vis) <- as(counts(vis), "sparseMatrix")
colnames(spatialCoords(vis)) <- c("x", "y")
接下来,我们执行 CARD 反卷积。在此,我们展示了 CARD 与 SingleCellExperiment 和 SpatialExperiment 的互操作性。反卷积结果矩阵已归一化,使得每个 spot 的细胞类型比例之和等于 1。
set.seed(2025)
CARD_obj <- CARD_deconvolution(
spe = vis,
sce = sce,
sc_count = NULL,
sc_meta = NULL,
spatial_count = NULL,
spatial_location = NULL,
ct_varname = "Annogrp",
ct_select = NULL, # decon with all sce$Annogrp cell types
sample_varname = NULL, # use all sce as one ref sample
mincountgene = 100,
mincountspot = 5
)
ws_card <- CARD_obj$Proportion_CARD
# order cell type names alphabetically, as for RCTD
ws_card <- data.frame(ws_card[, colnames(ws_rctd)])
ws_card[1:5, 1:5] |> round(2)
## B DCIS1 DCIS2 T dendritic
## AACACCTACTATCGAA-1 0 0 0 0.00 0
## AACACGTGCATCGCAC-1 0 0 0 0.00 0
## AACACTTGGCAAGGAA-1 0 0 0 0.01 0
## AACAGGAAGAGCATAG-1 0 0 0 0.02 0
## AACAGGATTCATAGTT-1 0 0 0 0.00 0
可视化
首先,我们定义了几个附件功能。
.plt_xy <- \(ws, vis, col, point_size) {
xy <- spatialCoords(vis)[rownames(ws), ]
colnames(xy) <- c("x", "y")
df <- cbind(ws, xy)
ggplot(df, aes(x, y, col=.data[[col]])) +
coord_equal() + theme_void() +
geom_point(size = point_size)
}
.plt_decon <- \(ws, vis){
lapply(names(ws), \(.)
.plt_xy(ws, vis, col = ., point_size = 0.3)) |>
wrap_plots(nrow = 3) & theme(
legend.key.width = unit(0.5, "lines"),
legend.key.height = unit(1, "lines")) &
scale_color_gradientn(colors = pals::jet())
}
我们可以在 x-y 空间中对反卷积权重进行可视化,即按照估计落入某个 spot 的给定细胞类型的比例进行着色:
.plt_decon(ws = ws_rctd, vis)
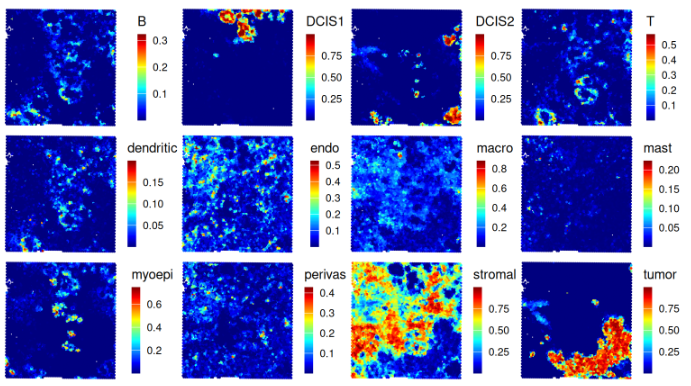
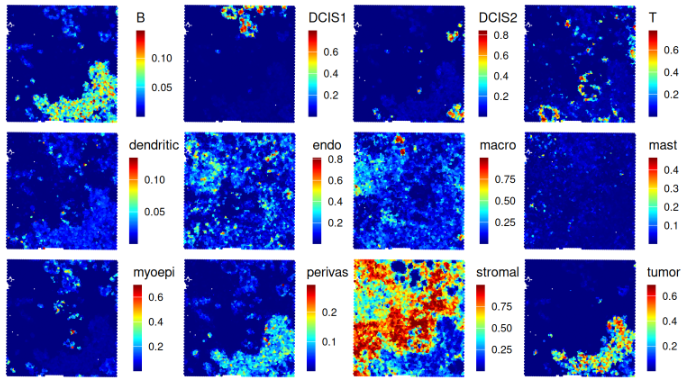
.plt_decon(ws = ws_card, vis)
反卷积结果也可以以热图形式查看,其中行 = 细胞,列 = 聚类:
plot_heat_ws <- function(ws, string){
p <- pheatmap(ws, show_rownames = FALSE, show_colnames = TRUE, main=string,
cellwidth = 12, treeheight_row = 5, treeheight_col = 5)
return(p)
}
plot_heat_ws(ws_rctd, string="RCTD")
plot_heat_ws(ws_card, string="CARD")
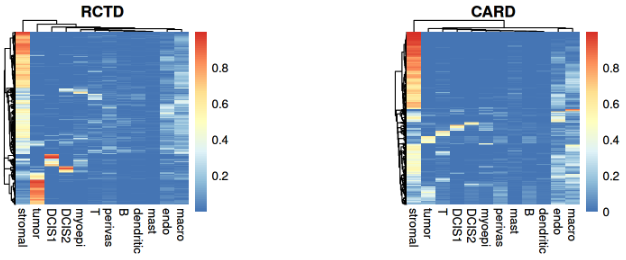
在两种方法中,我们都观察到超过一半的 spot 被估计具有超过 50 % 的 stromal 比例。少数 spot 对癌性子群体 DCIS1 和 DCIS2 表现出强烈且独特的信号。
为了与 10x Genomics 提供的 spot 注释进行比较,我们纳入了由 RCTD 反卷积得出的细胞类型。
ws <- ws_rctd
# derive majority vote label
ids <- names(ws)[apply(ws, 1, which.max)]
names(ids) <- rownames(ws)
vis$RCTD <- factor(ids[colnames(vis)])
# derive majority vote excluding stromal cells
ws_no_stroma <- ws[, colnames(ws) != "stromal"]
ids_no_stroma <- names(ws_no_stroma)[apply(ws_no_stroma, 1, which.max)]
names(ids_no_stroma) <- rownames(ws)
vis$RCTD_no_stroma <- factor(ids_no_stroma[colnames(vis)])
我们可以在空间上可视化这三个注释:
lapply(c("anno", "RCTD", "RCTD_no_stroma"),
\(.) plotCoords(vis, annotate=.)) |>
wrap_plots(nrow = 1) &
theme(legend.key.size = unit(0, "lines")) &
scale_color_manual(values = unname(pals::trubetskoy()))
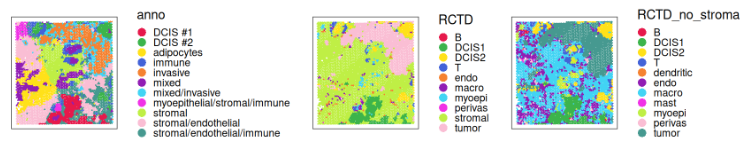
请注意强烈的 stromal 信号,以及 macrophages 是 stromal 细胞中第二常见的细胞类型。为了帮助表征来自反卷积的亚群,我们可以将它们的空间分布与提供的注释进行对比查看:
cd <- data.frame(colData(vis))
df <- as.data.frame(with(cd, table(RCTD, anno)))
fd <- as.data.frame(with(cd, table(RCTD_no_stroma, anno)))
ggplot(df, aes(Freq, RCTD, fill = anno)) + ggtitle("RCTD") +
ggplot(fd, aes(Freq, RCTD_no_stroma, fill = anno)) + ggtitle("RCTD_no_stroma") +
plot_layout(nrow = 1, guides = "collect") &
labs(x = "Proportion", y = NULL) &
coord_cartesian(expand = FALSE) &
geom_col(width = 1, col = "white", position = "fill") &
scale_fill_manual(values = unname(pals::trubetskoy())) &
theme_minimal() & theme(aspect.ratio = 1,
legend.key.size = unit(2/3, "lines"),
plot.title = element_text(hjust=0.5))
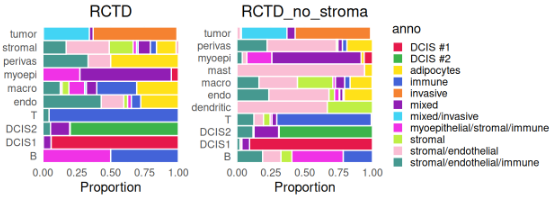
接下来,我们可以探究所提供的注释与两种反卷积标签之间的一致性。
hm <- \(mat, string) pheatmap(
mat, show_rownames = TRUE, show_colnames = TRUE, main=string,
cellwidth = 10, cellheight = 10, treeheight_row = 5, treeheight_col = 5)
hm(prop.table(table(vis$anno, vis$RCTD), 2), string="RCTD")
hm(prop.table(table(vis$anno, vis$RCTD_no_stroma), 2), string="RCTD_no_stroma")
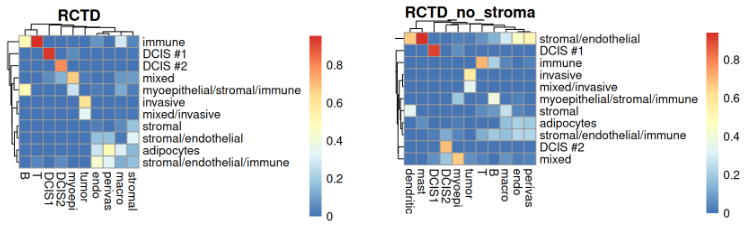
总体而言,我们观察到所提供的 spot 标签与 RCTD 反卷积得出的注释之间存在一致性。
在清除 stromal 之前,某些免疫细胞类型(如 dendritic 和 mast)从未有机会获得最高的细胞类型比例。在左图中,在 RCTD 注释为 T 细胞的所有 spot 中,几乎所有这些 spot 在提供的注释中都属于 “immune” 类型。对于标注为 “DCIS1”、“DCIS2” 和 “Invasive tumor” 的 spot,也观察到高度一致。
总之,基于反卷积的细胞类型比例估计能够重现 PCs,进而重现表达变异性。
未完待续,欢迎关注!
Reference
[1]
Ref: https://lmweber.org/OSTA/
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-09-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号