《python单细胞最佳实践》——归一化部分详细解读


书接上回,我们分享了:《单细胞最佳实践》—重新回顾质控部分的新理解,并对其中提到的资源进行了大力分享:python单细胞学习:超详细的单细胞最佳实践,初学或者升级技能都合适。
如果你也想打开学习,可以加微信:Biotree123拉你进群共同打开学习上面这个python版本的单细胞数据实践。
2.1 Motivation
上一节中,我们已经从数据集中移除了低质量细胞、环境 RNA 污染和 doublets,数据是 cells x genes 形式的计数矩阵(count matrix)。这些 count 代表了单细胞 RNA 测序实验中一个分子的捕获、反转录和测序,这些步骤中的每一步都会给同一细胞的测量计数深度引入一定的变异性,所以细胞间基因表达的差异可能仅仅是由于抽样效应(sampling effects)造成的。这意味着计数矩阵仍然包含差异很大的方差项。分析数据集通常具有挑战性,因为许多统计方法假设数据具有均匀的方差结构。
伽马-泊松分布 (Gamma-Poisson distribution) 一个在理论和经验上均适用于 UMI 数据的模型是伽马-泊松分布。该分布有一个二次均值-方差关系式:方差 μαμ,其中 μ 是均值,α 是过离散(overdispersion)参数。当 α=0 时,即为泊松分布;α>0 描述了超出泊松分布的额外方差。
“标准化”这一预处理步骤旨在通过将可观测的方差调整到指定范围,来校正数据集中原始 count 因可变抽样效应带来的影响。实践中使用了几种复杂程度各不相同的标准化技术,它们的设计通常是为了确保后续分析任务以及基础统计方法能够适用。
《Comparison of transformations for single-cell RNA-seq data》这项基准研究比较了单细胞数据的 22 种不同转换方法。它基于细胞与 ground truth 之间的图重叠(graph overlap)比较不同标准化技术的性能。我们需要强调的是,一个完整的基准测试还需要比较标准化对各种不同下游分析任务的影响,这仍有待完成。我们建议分析人员谨慎选择标准化方法,并始终考虑后续的分析任务。
本章将向读者介绍三种不同的标准化技术:移位对数变换(the shifted logarithm transformation)、scran 标准化(scran normalization)、分析性 Pearson 残差近似(analytic approximation of Pearson residuals)。移位对数变换有利于稳定方差,适用于后续降维和差异表达基因的识别;scran 经过广泛测试并用于批次校正任务;pearson 残差适用于选择生物可变基因和识别稀有细胞类型。
首先导入 python 包和已经按照上一章内容进行质控的数据集。
import logging
import anndata2ri
import rpy2.rinterface_lib.callbacks as rcb
import rpy2.robjects as ro
import scanpy as sc
import seaborn as sns
from matplotlib import pyplot as plt
from scipy.sparse import issparse
sc.settings.verbosity = 0
sc.settings.set_figure_params(
dpi=80,
facecolor="white",
# color_map="YlGnBu",
frameon=False,
)
rcb.logger.setLevel(logging.ERROR)
# ro.pandas2ri.activate() # activate()方法已被弃用(deprecated),改用上下文管理器(context manager)
# anndata2ri.activate()
converter = ro.conversion.Converter("my_converter")
converter = ro.pandas2ri.converter + converter
converter = anndata2ri.converter + converter
with ro.conversion.localconverter(converter):
%load_ext rpy2.ipython
adata = sc.read(
filename="s4d8_quality_control.h5ad",
backup_url="https://figshare.com/ndownloader/files/40014331",
)
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False)
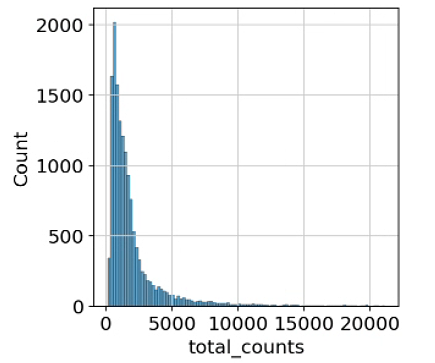
2.2 移位对数变换
移位对数变换是基于 delta 的方法。delta 方法对原始计数 应用一个非线性函数 ,旨在使整个数据集中的方差更加相似。
移位对数变换通过以下方法实现:
其中 是原始计数, 是所谓的尺寸因子(size factor),通过来校正技术偏差(测序深度), 是一个伪计数(pseudo-count)。为每个细胞确定尺寸因子,以考虑抽样效应和不同细胞大小的变化。细胞的尺寸因子 可以通过以下公式计算:
其中 代表不同基因的索引, 被描述为 a target sum(scanpy.pp.normalize_total()中的target_sum参数),这是一个标准化常数或缩放因子。存在多种不同的方法可以从数据中确定尺寸因子。在本节中,我们将利用 scanpy 的默认设置,即为数据集中原始计数深度(raw count depth)的中位数。许多分析模板使用固定的值,比如,这会产生通常称为 CPM(counts per million)的值。For a beginner, these values may seem arbitrary, but it can lead to much larger overdispersions than typically seen in single-cell datasets. 对于初学者而言,这些值(指的应该是固定值,如 CPM 标准化结果)可能是随意的,但它可以导致比通常情况更大的过度离散(单细胞数据本身存在过度离散,但 CPM 会人为制造更极端的过度离散,CPM 的L=1e6来自 bulk RNA-seq 的约定俗成,没有考虑到单细胞数据总计数普遍较低的特性(中位数可能仅几万),因此 seurat 中的默认参数是L=1e4)。
过度离散(Overdispersion) 过度离散描述了数据集中存在的变异性大于预期的情况(可以理解为基因表达方差远大于均值,不符合泊松分布,超出的方差称为过度离散)。
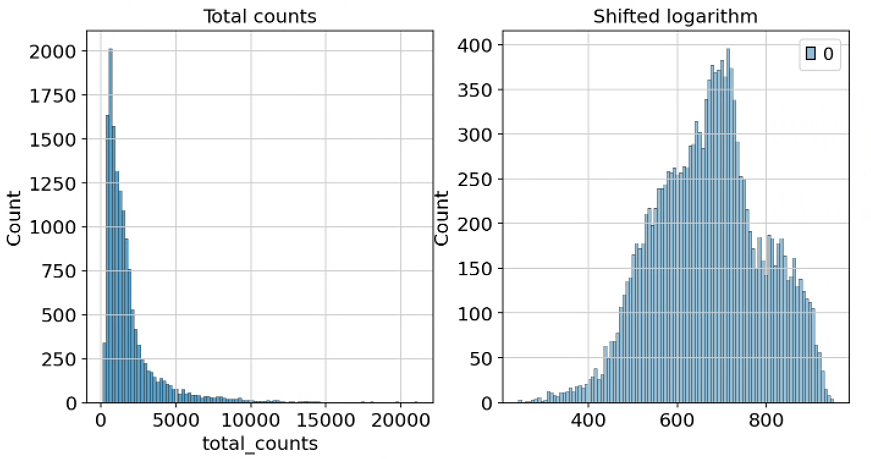
移位对数变换是一种快速的标准化技术,在揭示数据集的潜在结构方面(尤其是在主成分分析之后)优于其他方法,并且有利于稳定方差以进行后续的降维和差异表达基因识别。在 scanpy 中,可以通过运行 pp.normalize_total 并设置 target_sum=None 来方便地调用移位对数变换。我们将 inplace 参数设置为 False,因为在本教程中我们想探索三种不同的标准化技术。第二步使用 scaled count,我们得到了第一个标准化计数矩阵。
scales_counts = sc.pp.normalize_total(adata, target_sum=None, inplace=False)
# log1p transform
adata.layers["log1p_norm"] = sc.pp.log1p(scales_counts["X"], copy=True)
可以比较应用移位对数变换前后 count 的分布变化:
fig, axes = plt.subplots(1, 2, figsize=(10, 5))
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False, ax=axes[0])
axes[0].set_title("Total counts")
p2 = sns.histplot(adata.layers["log1p_norm"].sum(1), bins=100, kde=False, ax=axes[1])
axes[1].set_title("Shifted logarithm")
plt.show()
2.3 scran 标准化
scran 标准化同样也是基于 delta 方法,是一种基于池化(pooling-based)的尺寸因子估计方法。scran 遵循与移位对数变换相同的原则,计算 ,唯一的区别是 scran 利用反卷积方法(deconvolution approach),基于对细胞池(pools of cells)的基因进行线性回归来估计尺寸因子。
如何理解?
传统方法(如总计数中位数)的缺陷:如果细胞群体包含不同细胞类型,A 类型细胞的总计数 天然较高, 较大;B 类型细胞的总计数天然较低, 较小,此时用 标准化就会错误地将真实的生物差异(总 RNA 含量不同)当作技术偏差校正掉,导致 B 类型细胞的所有基因表达被人为“拉高”。
反卷积法的思想:将细胞随机分成多个小池(pool),每个池包含几十到几百个细胞。对每个基因,在该池的所有细胞上拟合一个线性模型,然后从所有池的回归结果中解耦(deconvolve)出每个细胞的独立尺寸因子。本质上,该方法试图在排除基因表达异质性影响的前提下,估计细胞的技术偏差。
scran 在批次校正任务中经过了广泛测试,可以通过相应的 R 包轻松调用。
%%R
library(scran)
library(BiocParallel)
scran 需要一个粗略的聚类输入以提高尺寸因子估计的性能。在本教程中,我们使用一种简单的预处理方法,以低分辨率对数据进行聚类,为尺寸因子估计提供输入。
# Preliminary clustering for differentiated normalization
adata_pp = adata.copy()
sc.pp.normalize_total(adata_pp)
sc.pp.log1p(adata_pp)
sc.pp.pca(adata_pp, n_comps=15)
sc.pp.neighbors(adata_pp)
sc.tl.leiden(adata_pp, key_added="groups")
现在我们将 data_mat 和聚类分组添加到 R 环境中。
data_mat = adata_pp.X.T
if issparse(data_mat):
if data_mat.nnz > 2**31 - 1:
data_mat = data_mat.tocoo()
else:
data_mat = data_mat.tocsc()
# ro.globalenv["data_mat"] = data_mat
# ro.globalenv["input_groups"] = adata_pp.obs["groups"]
注释掉的这两行是我跑的时候会报 NotImplementedError: Conversion 'py2rpy' not defined for objects of type '<class 'scipy.sparse._csc.csc_matrix'>' 和 NotImplementedError: Conversion 'py2rpy' not defined for objects of type '<class 'pandas.core.series.Series'>' 错误,下面是我成功跑出来的解决方法:
Matrix = ro.packages.importr('Matrix')
new = ro.r['new']
# 转换csc_matrix为R的dgCMatrix
def csc_to_dgCMatrix(csc_mat):
# 获取矩阵属性
dim = csc_mat.shape
i = csc_mat.indices.astype(int)
p = csc_mat.indptr.astype(int)
x = csc_mat.data.astype(float)
# 创建R的dgCMatrix对象
r_dgCMatrix = new(
Class="dgCMatrix",
i=ro.IntVector(i),
p=ro.IntVector(p),
x=ro.FloatVector(x),
Dim=ro.IntVector(dim)
)
return r_dgCMatrix
r_dgCMatrix = csc_to_dgCMatrix(data_mat)
ro.globalenv["data_mat"] = r_dgCMatrix
ro.globalenv["input_groups"] = ro.vectors.StrVector(adata_pp.obs["groups"].astype(str).tolist())
相当于手动转换成 R 能识别的格式,感觉这样笨笨的,应该会有更简单的办法,activate() 应该可以自动转换,但前面说这个方法已被弃用,后续如果有时间再看一下这里吧。
现在就可以删掉 adata_pp 了,因为我们已经获得了运行 scran 需要的所有对象。
del adata_pp
然后计算尺寸因子:
%%R -o size_factors
size_factors = sizeFactors(
computeSumFactors(
SingleCellExperiment(
list(counts=data_mat)
),
clusters = input_groups,
min.mean = 0.1,
BPPARAM = MulticoreParam()
)
)
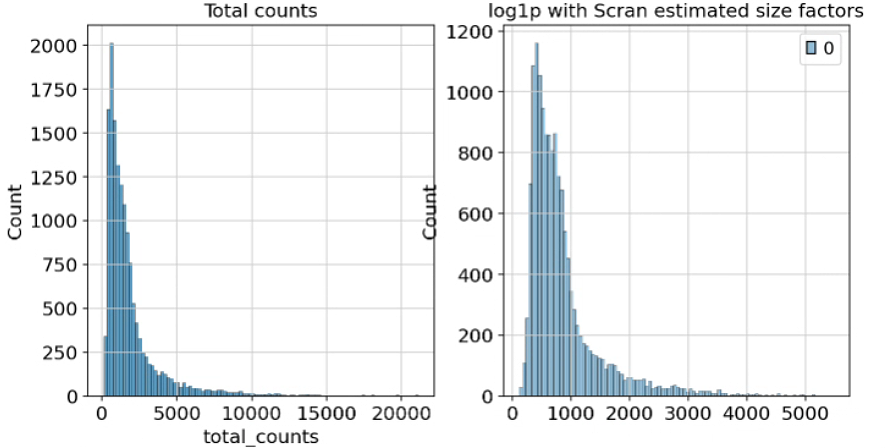
将 size_factors 保存在 .obs 中,现在能够标准化数据并随后应用 log1p 变换。
adata.obs["size_factors"] = size_factors
scran = adata.X / adata.obs["size_factors"].values[:, None]
adata.layers["scran_normalization"] = csr_matrix(sc.pp.log1p(scran))
fig, axes = plt.subplots(1, 2, figsize=(10, 5))
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False, ax=axes[0])
axes[0].set_title("Total counts")
p2 = sns.histplot(
adata.layers["scran_normalization"].sum(1), bins=100, kde=False, ax=axes[1]
)
axes[1].set_title("log1p with Scran estimated size factors")
plt.show()
2.4 Pearson 残差
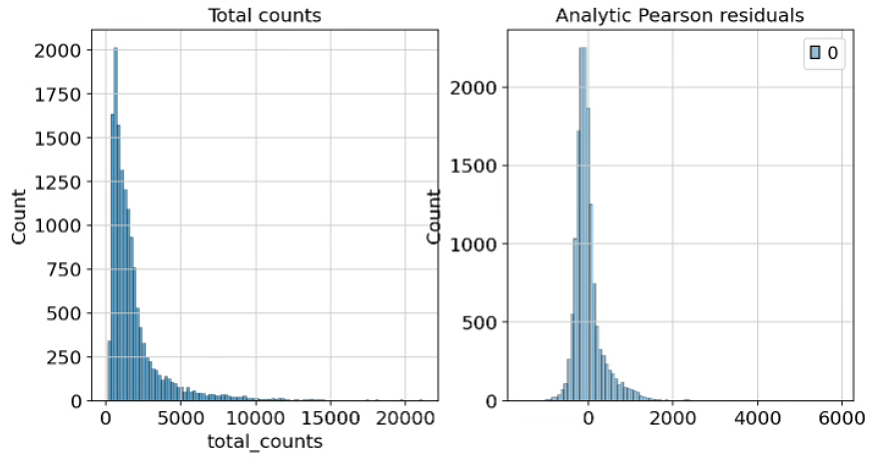
我们在本章介绍的第三种归一分析性 Pearson 残差近似。这种标准化技术的动机源于观察到 scRNA-seq 数据中的细胞间变异可能受到生物异质性(biological heterogeneity)与技术效应(technical effects)的混杂影响。该方法利用“正则化负二项回归(regularized negative binomial regression)”得到的 Pearson 残差(Pearson residuals)来计算数据中技术噪音的模型。它明确地将计数深度作为协变量添加到广义线性模型中。《pipeComp, a general framework for the evaluation of computational pipelines, reveals performant single cell RNA-seq preprocessing tools》这篇文章在不同标准化技术的独立比较中表明,该方法在保留数据集细胞异质性的同时消除了抽样效应的影响。值得注意的是,分析性 Pearson 残差不需要下游的启发式步骤,如添加伪计数或对数转换。该方法的输出是标准化的值,可以是正数或负数。一个细胞和基因的负残差表明,与该基因的平均表达量和细胞测序深度相比,观察到的计数少于预期。正残差则表明计数多于预期。分析性 Pearson 残差在 scanpy 中实现,可以直接在原始计数矩阵上计算。
analytic_pearson = sc.experimental.pp.normalize_pearson_residuals(adata, inplace=False)
adata.layers["analytic_pearson_residuals"] = csr_matrix(analytic_pearson["X"])
fig, axes = plt.subplots(1, 2, figsize=(10, 5))
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False, ax=axes[0])
axes[0].set_title("Total counts")
p2 = sns.histplot(
adata.layers["analytic_pearson_residuals"].sum(1), bins=100, kde=False, ax=axes[1]
)
axes[1].set_title("Analytic Pearson residuals")
plt.show()
我们将不同的标准化技术应用于数据集,并将它们作为单独的层保存到我们的 anndata 对象中。根据后续的分析任务,使用不同标准化的层并评估结果可能是有利的。
adata.write("s4d8_normalization.h5ad")本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-08-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号