inferCNVpy分析流程学习
原创

inferCNV的用途大家都已经很清楚了,接下来就走一下流程,试一试Python版的inferCNV。
使用的数据集为:GSE188711,既往推文可见:Scanpy分析全流程(含harmonypy整合/细胞周期矫正/双细胞检测及去除)(https://mp.weixin.qq.com/s/qQ5gq_yvOzu4n1pcL6NMYw)
分析流程
1. 安装
conda activate sc
pip install inferCNVpy2. 导入
# 如果没有安装请提前安装哦
import scanpy as sc
import infercnvpy as cnv
import matplotlib.pyplot as plt
import warnings
warnings.simplefilter("ignore")
sc.settings.set_figure_params(figsize=(5, 5))# check一下umap
sc.pl.umap(adata, color="celltype")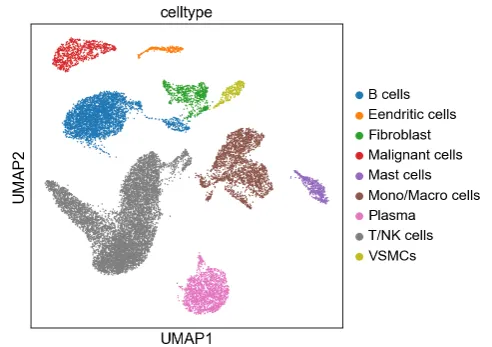
3. 下载并处理基因组文件
示例文件中将基因组信息分别按照ensg,chromosome,start和end命名整合到了adata.var中去了。
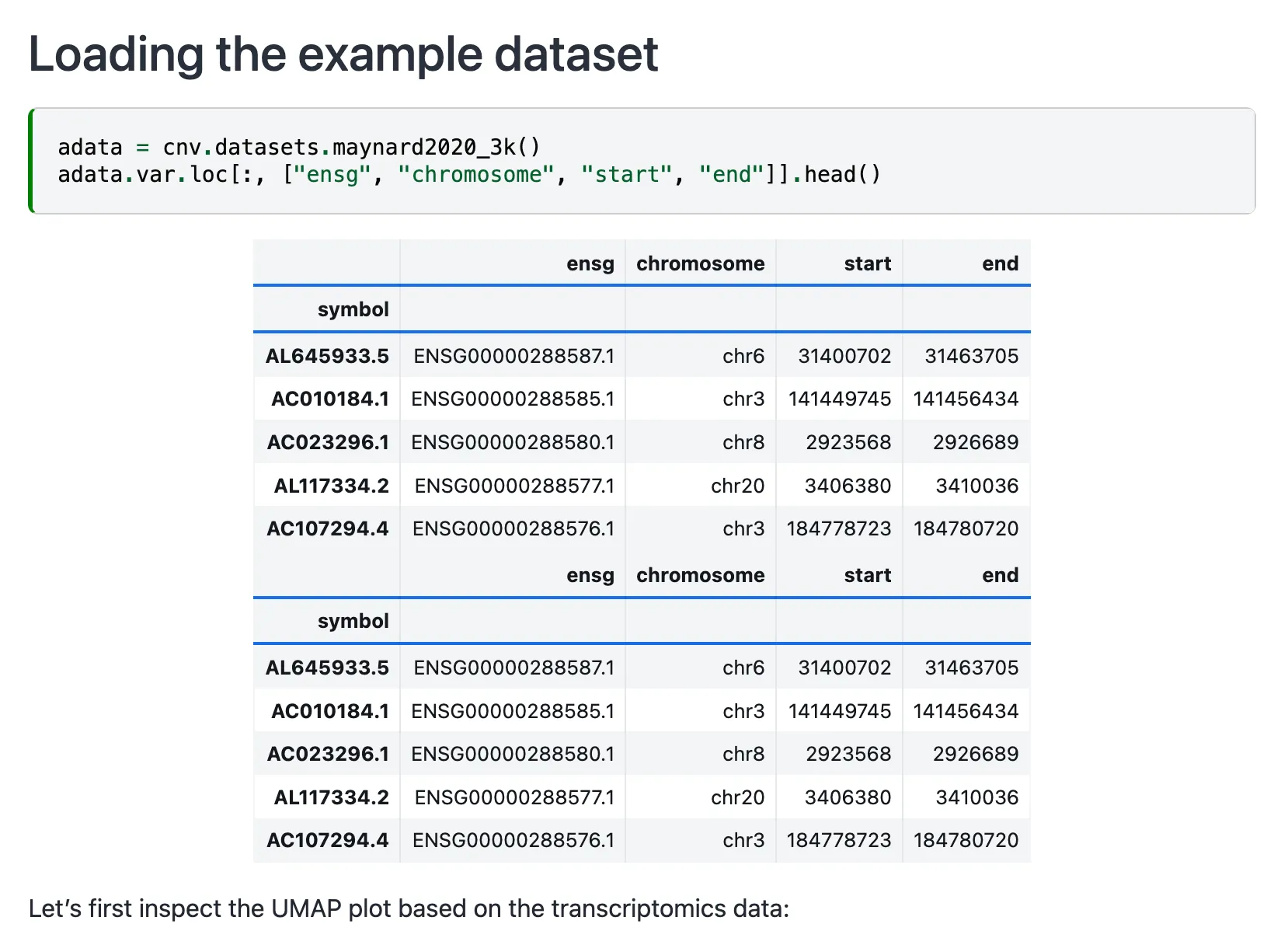
因此还需要提前把基因组注射文件下载好(https://data.broadinstitute.org/Trinity/CTAT/cnv/),要分清不同物种

# 在终端中下载
wget https://data.broadinstitute.org/Trinity/CTAT/cnv/hg38_gencode_v27.txt# 读取基因组注释文件
# 自定义列名(请根据实际列数填写)
column_names = ["gene_symbol", "chromosome", "start", "end"] # 示例字段,可根据实际调整
anno = pd.read_csv(
"hg38_gencode_v27.txt",
delim_whitespace=True,
engine="python",
header=None,
names=column_names
)
anno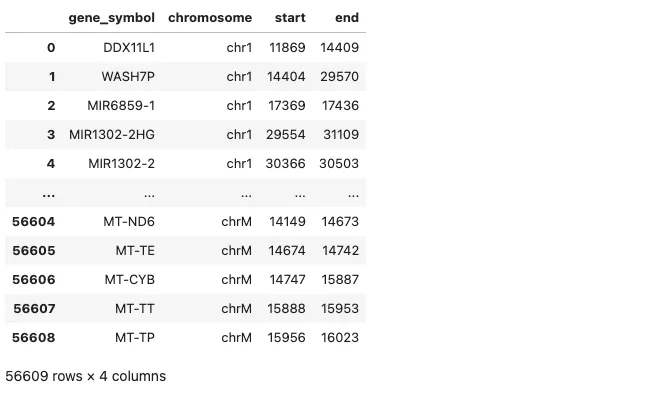
4. 接下来把gene_symbol与adata的基因名对应,然后把chromoseme等信息对应上去
# 确保 anno 以 gene_symbol 为索引
anno_indexed = anno.set_index("gene_symbol")
# 执行 left join:以 adata.var 的 index 为主(即 adata.var_names)
adata.var = adata.var.join(anno_indexed[["chromosome", "start", "end"]], how="left")
adata.var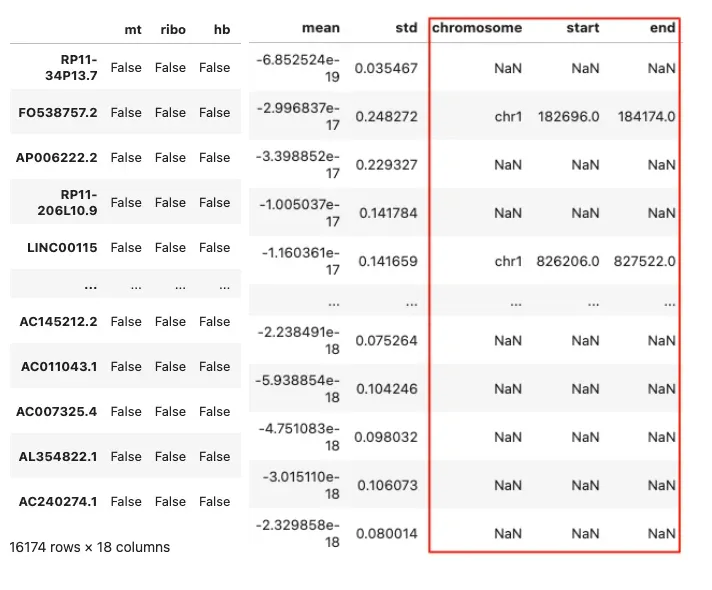
# 检查是否存在重复基因名称,需要去除
duplicates = adata.var_names[adata.var_names.duplicated()]
print(duplicates)
#Index([], dtype='object')#还可以看一下有哪些celltype
adata.obs["celltype"].value_counts()
#celltype
#T/NK cells 8917
#B cells 3685
#Mono/Macro cells 2502
#Plasma 1774
#Malignant cells 809
#Fibroblast 698
#Mast cells 477
#VSMCs 290
#Eendritic cells 258
#Name: count, dtype: int645.正式开始分析,官网使用了所有免疫细胞作为参照
# 官网使用了所有的免疫细胞作为reference细胞
cnv.tl.infercnv(
adata,
reference_key="celltype",
reference_cat=[
"T/NK cells",
"B cells",
"Mono/Macro cells",
"Mast cells",
],
window_size=250,
)
# 警示中提示了多少基因是没有匹配上的,同时可以看一下运行时间,非常快速!
# WARNING: Skipped 1581 genes because they don't have a genomic position annotated.
# 100%|█████████████████████████████████████████████████| 4/4 [00:14<00:00, 3.59s/it]cnv.pl.chromosome_heatmap(adata, groupby="celltype")但感觉这个数据集中CNV的差异并不显著
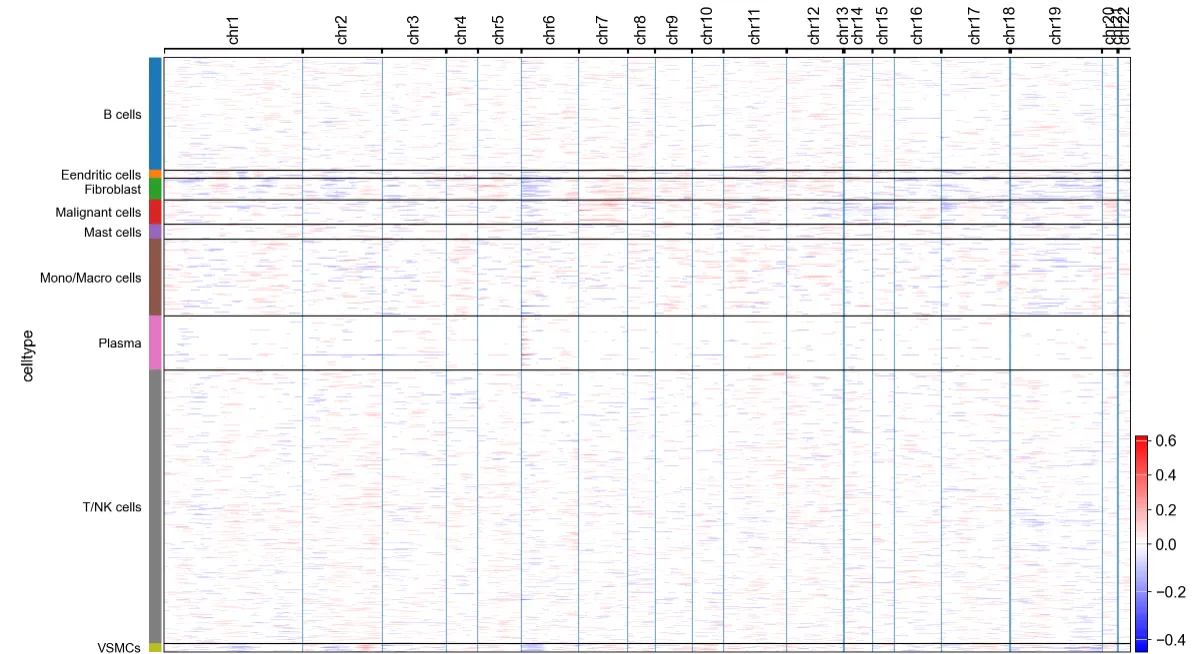
6.根据CNV特征进行聚类并识别肿瘤细胞(官网提供的配套分析流程)
cnv.tl.pca(adata)
cnv.pp.neighbors(adata)
cnv.tl.leiden(adata)cnv.pl.chromosome_heatmap(adata, groupby="cnv_leiden", dendrogram=True)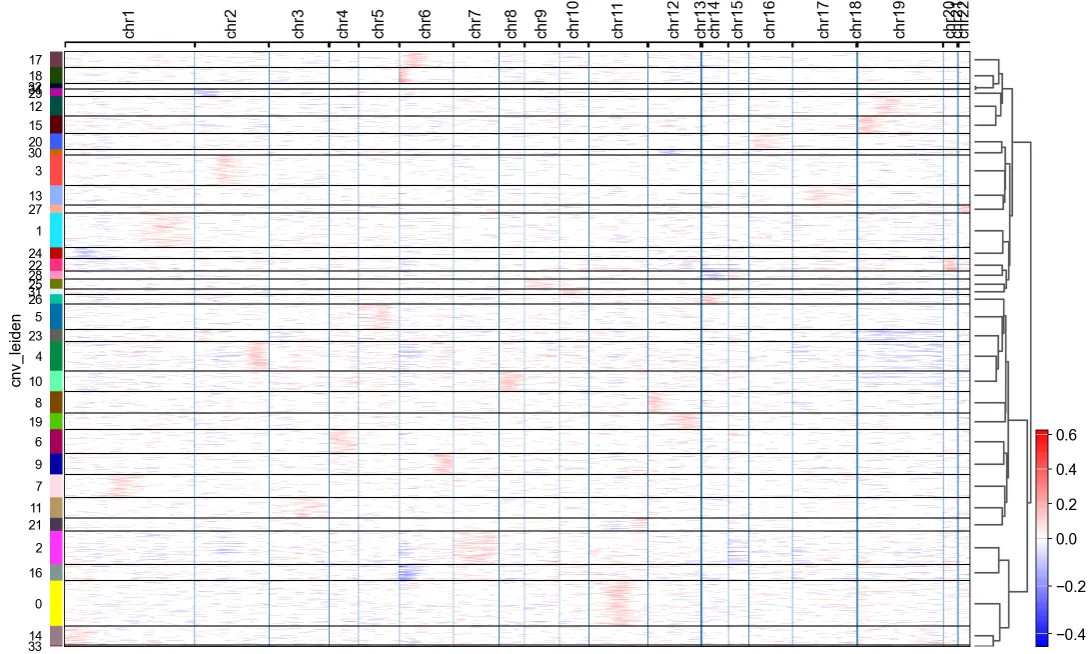
7.根据CNV特征所构建的UMAP图谱(官网提供的配套分析流程)
cnv.tl.umap(adata)
cnv.tl.cnv_score(adata)fig, ((ax1, ax2), (ax3, ax4)) = plt.subplots(2, 2, figsize=(11, 11))
ax4.axis("off")
cnv.pl.umap(
adata,
color="cnv_leiden",
legend_loc="on data",
legend_fontoutline=2,
ax=ax1,
show=False,
)
cnv.pl.umap(adata, color="cnv_score", ax=ax2, show=False)
cnv.pl.umap(adata, color="celltype", ax=ax3)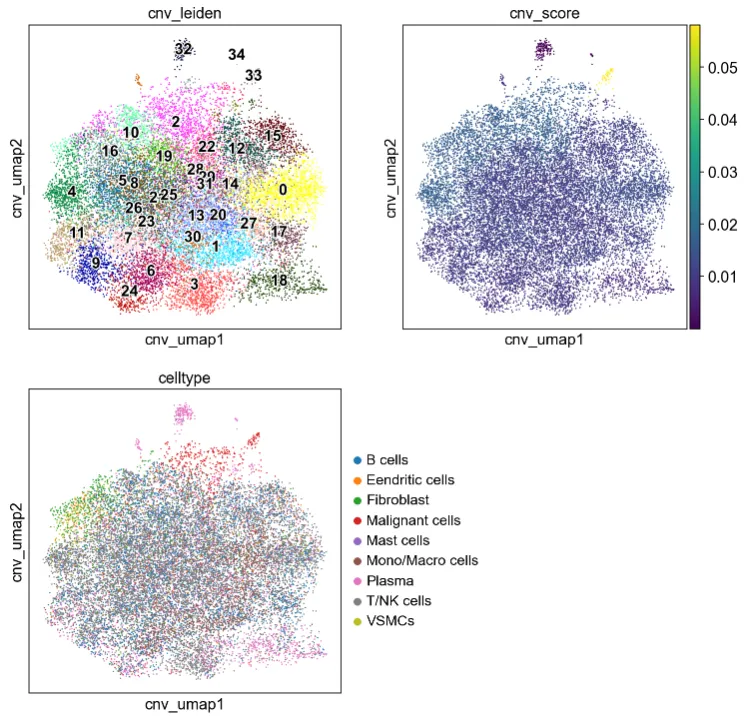
8.按照CNV情况对肿瘤细胞进行分类(官网提供的配套分析流程)
# 按照CNV_score分数第33簇细胞的CNV分数较高,设定为肿瘤细胞
adata.obs["cnv_status"] = "normal"
adata.obs.loc[adata.obs["cnv_leiden"].isin(["33"]), "cnv_status"] = (
"tumor"
)fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(12, 5), gridspec_kw={"wspace": 0.5})
cnv.pl.umap(adata, color="cnv_status", ax=ax1, show=False)
sc.pl.umap(adata, color="cnv_status", ax=ax2)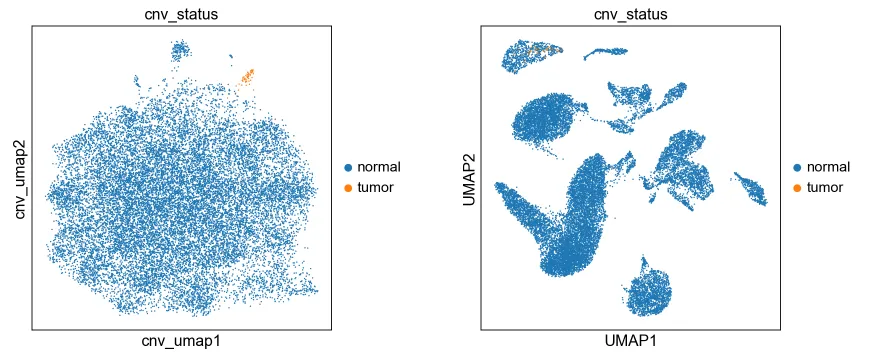
# 单独显示tumor细胞的CNV热图
cnv.pl.chromosome_heatmap(adata[adata.obs["cnv_status"] == "tumor", :])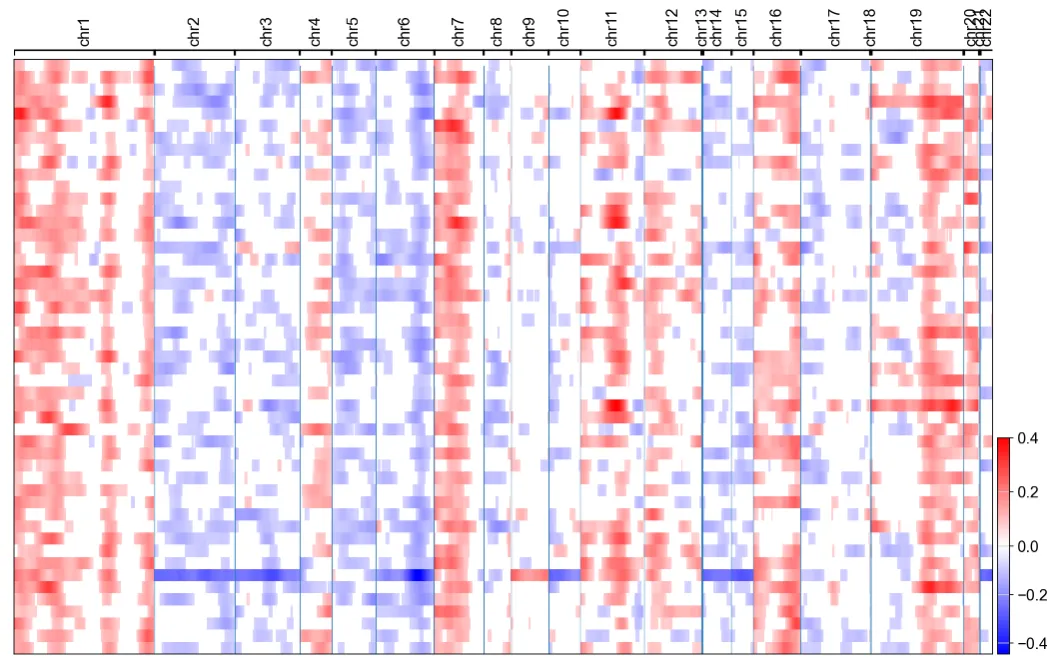
# 单独显示normal细胞的CNV热图
cnv.pl.chromosome_heatmap(adata[adata.obs["cnv_status"] == "normal", :])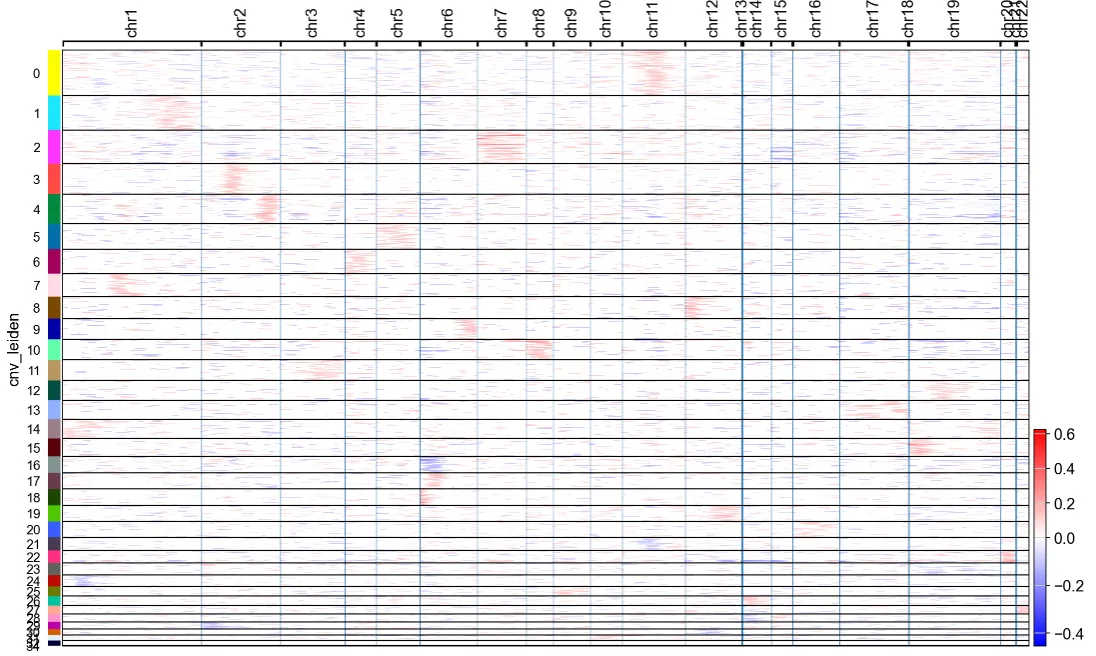
参考资料
- inferCNVpy github: https://github.com/icbi-lab/infercnvpy
- 生信技能树:https://mp.weixin.qq.com/s/dcPYo9eR2CaPCr1-MbyC6w
- 单细胞天地:https://mp.weixin.qq.com/s/y1Q_GGvVD-eHtrqg_rkKTg
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多相关内容可关注公众号:生信方舟
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号