做单细胞和空转必须了解的AnnData数据结构


今天来学习如何对 python中使用非常多的 AnnData 对象进行数据提取,绘图操作。AnnData 对象几乎可以匹配R语言中单细胞数据分析的Seurat对象,了解其每一个元素有利于我们更加灵活的做各种分析~
图解如下:https://anndata.readthedocs.io/en/stable/index.html
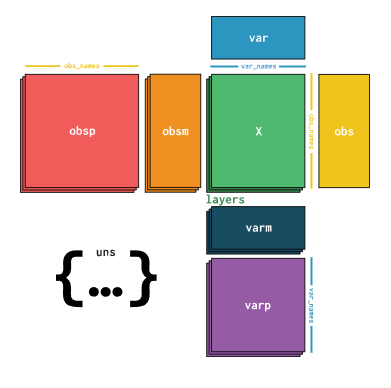
0.示例数据
既然是做单细胞和空转必须了解的对象,我们这里就使用单细胞转录组数据 GSE163558 作为参考,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163558。
GSE163558数据集共10个样本,下载 GSE163558_RAW.tar,处理成下面的格式:
GSE163558
├── GSM5004180_PT1
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── GSM5004181_PT2
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── GSM5004182_PT3
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── GSM5004183_NT1
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
...
1.数据读取
首先加载读取所需要的模块:
import pandas as pd
import scanpy as sc
import numpy as np
import os
# 列出当前目录中的样本
dir = os.listdir("GSE163558_RAW")
dir
输出结果如下,[]表示数据是一个列表:
['GSM5004180_PT1',
'GSM5004181_PT2',
'GSM5004182_PT3',
'GSM5004183_NT1',
'GSM5004184_LN1',
'GSM5004185_LN2',
'GSM5004186_O1',
'GSM5004187_P1',
'GSM5004188_Li1',
'GSM5004189_Li2']
现在批量读取,并将多个对象合并在一起,使用for循环:
# for循环读取:
adata = {}
for i in range(len(dir)):
data = sc.read_10x_mtx("GSE163558_RAW/" +dir[i], var_names="gene_symbols", cache=True)
data.var_names_make_unique()
adata[dir[i]] = data
print(dir[i])
# 使用 concat 函数将多个adata连接在一起:
adata = sc.concat(adata,label='sampleid')
adata.obs_names_make_unique()
adata
输出对象:现在是一个只有54687个细胞,33538个基因的初始AnnData对象,obs为每一个细胞所属的样本
AnnData object with n_obs × n_vars = 54687 × 33538
obs: 'sampleid'
简单探索一下,如下面的操作:
# 每个样本里的细胞数量
adata.obs.sample.value_counts()
# 表达矩阵里的数值范围
np.min(adata.X), np.max(adata.X)
# 基本过滤
# 过滤前 的细胞数与基因数
adata.X.shape
# (54687, 33538)
# 每个细胞中表达多少个基因
adata.obs.head()
保存上面的每个样本里的细胞数为一个csv文件:
temp = adata.obs.sampleid.value_counts()
# 用type()获取对象的数据类型
type(temp)
# pandas.core.series.Series
# 保存为CSV文件
temp.to_csv('sampleid_counts.csv', header=True, index=True)
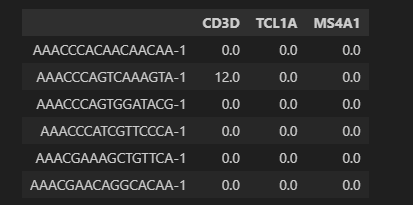
查看感兴趣的基因的表达矩阵:稀疏矩阵不支持直接查看,只能是转换成矩阵或者数据框才能查看。转换成矩阵就丢失了行名列名,转换成数据框更好。
# 转换成矩阵
adata[0:6, ['CD3D','TCL1A','MS4A1']].X.toarray()
# 转换成数据框
adata[0:6, ['CD3D','TCL1A','MS4A1']].to_df()
2.数据质控
这一步主要对低表达基因以及低质量细胞进行过滤。
首先计算每个细胞中线粒体基因,核糖体基因,红血细胞基因表达比例:
# 计算线粒体基因比例
adata.var['mt']=adata.var_names.str.startswith('MT-')
# 计算核糖体基因比例
adata.var['ribo']=adata.var_names.str.match('^RP[SL]')
# 计算红血细胞基因比例
adata.var['hb']=adata.var_names.str.match('^HB[^(P)]')
# 计算
sc.pp.calculate_qc_metrics(adata,qc_vars=['mt','ribo','hb'],percent_top=None,log1p=False,inplace=True)
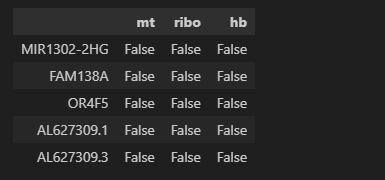
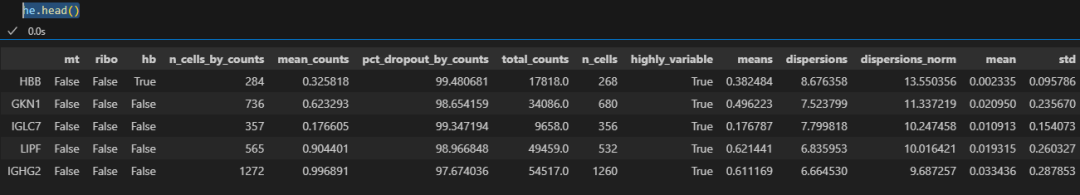
看每个基因:
# 每个基因在多少个细胞中表达
adata.var.head()
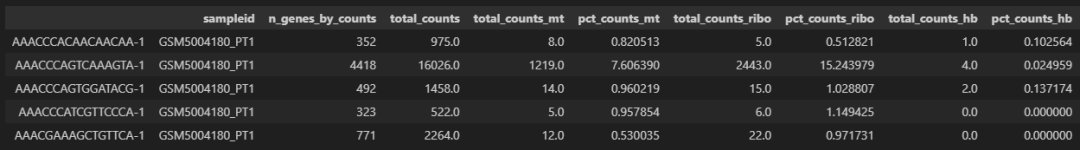
每个细胞中的指标:
# 每个细胞中表达多少个基因
adata.obs.head()
可视化上述常见的三个参数:
# 可视化细胞的上述比例情况
sc.pl.violin(adata,['n_genes_by_counts','total_counts','pct_counts_mt'],groupby='sampleid',jitter=False,rotation=90)
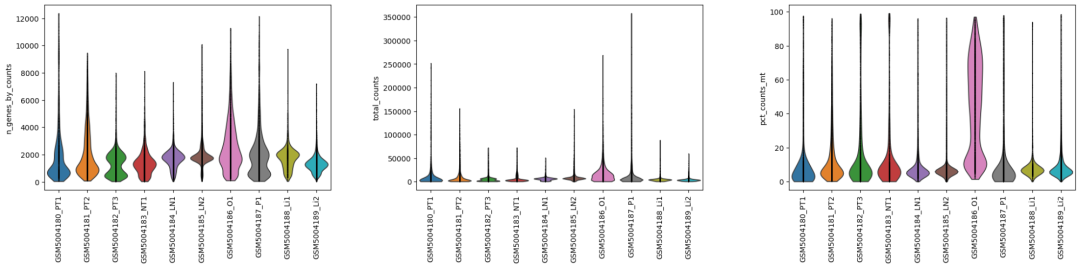
调整一些参数
jitter:可以设置散点抖动的宽度rotation:可以调整x轴标签的旋转角度,样本多可以设置90度size:设置散点的大小
# 可视化细胞的上述比例情况
sc.pl.violin(adata,['n_genes_by_counts','total_counts','pct_counts_mt'],groupby='sampleid',jitter=0.4,rotation=90,size=0.8)
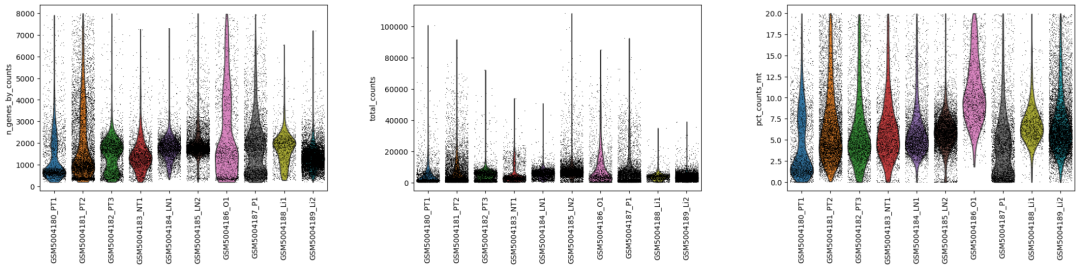
过滤:
# 过滤细胞,每个细胞中至少200个基因表达
sc.pp.filter_cells(adata,min_genes=200)
# 过滤基因,每个基因至少在5个细胞中表达
sc.pp.filter_genes(adata,min_cells=5)
# 过滤细胞:大于8000个基因的过滤
adata = adata[adata.obs.n_genes_by_counts < 8000, :]
# 过滤细胞:线粒体基因表达比例大于20%的过滤
adata = adata[adata.obs.pct_counts_mt < 20, :].copy()
adata
过滤完后还有:44238个细胞,26480个基因
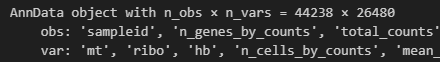
3.降维聚类
基本上都是scanpy的标准流程了:
# 首先将数据矩阵标准化(校正文库大小):
sc.pp.normalize_total(adata,target_sum=1e4)
# 对数据进行log
sc.pp.log1p(adata)
adata.raw = adata.copy()
# 高变基因
sc.pp.highly_variable_genes(adata)
# 归一化
sc.pp.scale(adata)
# pca分析
sc.tl.pca(adata,use_highly_variable=True)
# harmony整合
sc.external.pp.harmony_integrate(adata,'sampleid')
# 聚类
sc.pp.neighbors(adata,use_rep='X_pca_harmony',n_pcs=30)
sc.tl.umap(adata)
# Using the igraph implementation and a fixed number of iterations can be significantly faster, especially for larger datasets
sc.tl.leiden(adata, flavor="igraph", n_iterations=2,resolution=0.5)
到这里我们的数据基本分析都已经做完了,降维聚类分群,harmony去批次。
再来看一下数据的变化:
adata这个时候除了obs,var还多了很多其他的:
- obs:里面保存的每一个细胞对应的表型信息,对应Seurat里面的metadata
- var:对应的每一个基因的相关信息
- uns:对应的是字典类型的元素,保存了如hvg, umap等信息
- obsm:保存的空间坐标
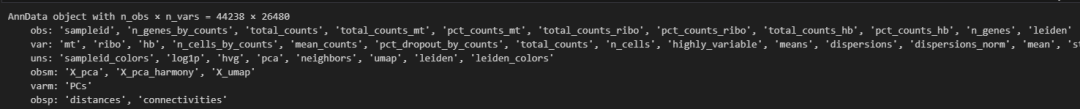
常规绘图
相关性:
total_counts 与 n_genes_by_counts的相关性:
# `total_counts` 与 `n_genes_by_counts`的相关性:
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts")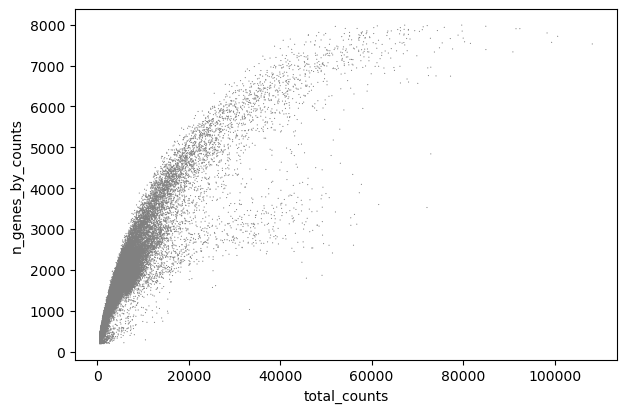
可视化高变基因:
sc.pl.highly_variable_genes(adata)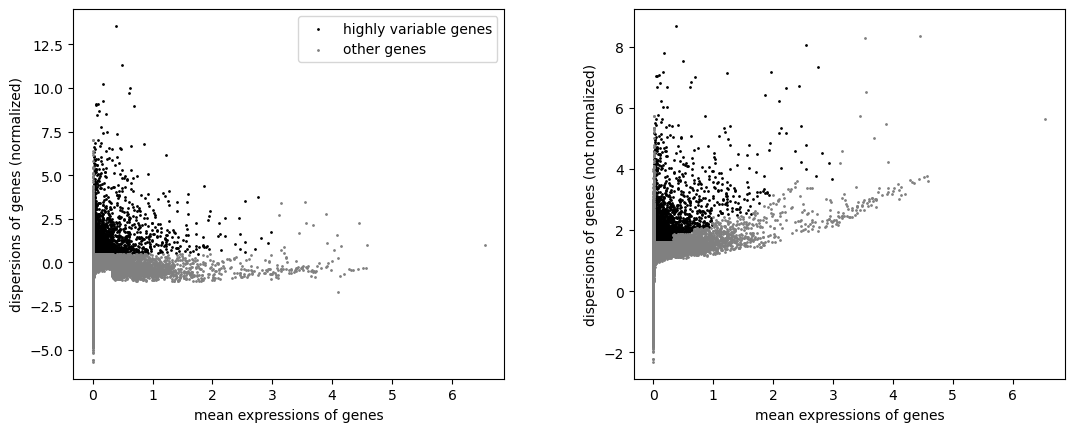
提取高变基因:
# adata.var.loc[adata.var.highly_variable == True]
he = adata.var.sort_values(by='dispersions_norm', ascending=False)
# pandas.core.frame.DataFrame对象
he.head()
# 提取高变基因
hvg = he[he.highly_variable == True].index.tolist()
len(hvg)
hvg[1:10]
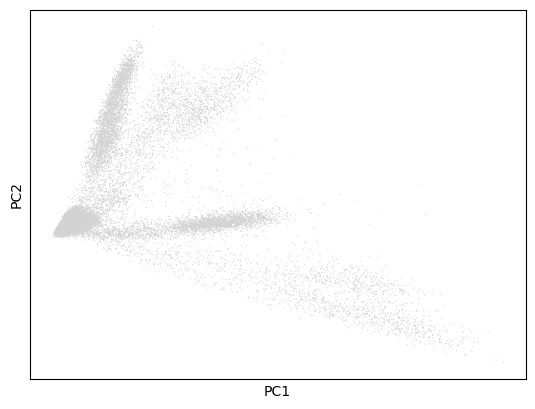
pca聚类结果:
# 绘制 pca 聚类结果
sc.pl.pca(adata)
更改一下颜色:
# 绘制 PCA 图,并根据 'sampleid' 列标记不同的样本
sc.pl.pca(adata, color='sampleid')
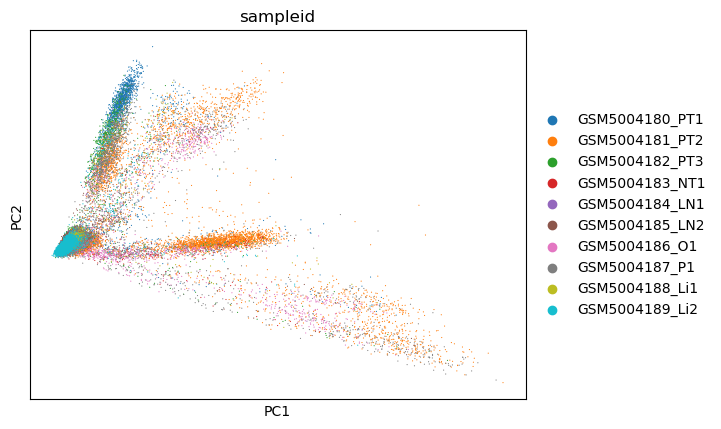
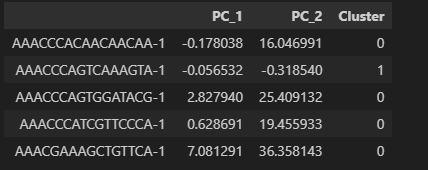
提取pca坐标自己绘图看看:
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
pca_df = pd.DataFrame({
'PC_1': adata.obsm['X_pca'][:, 0],
'PC_2': adata.obsm['X_pca'][:, 1],
'Cluster': adata.obs['leiden']
})
pca_df.head()
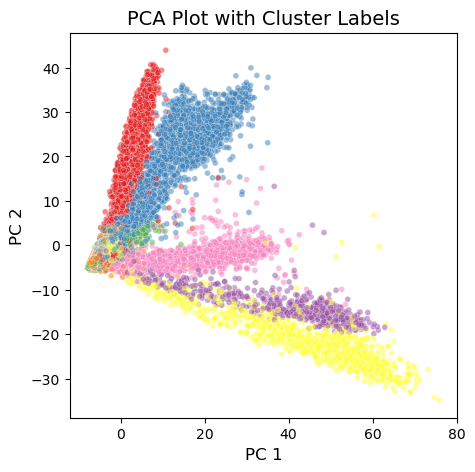
绘图:
plt.figure(figsize=(5, 5))
# 使用 seaborn 绘制散点图,按 Cluster 着色
sns.scatterplot(
data=pca_df,
x='PC_1',
y='PC_2',
hue='Cluster',
palette='Set1',
alpha=0.5,
size=0.1,
legend=None# 不显示默认图例
)
# 图片设置
plt.grid(False)
plt.title('PCA Plot with Cluster Labels', fontsize=14)
plt.xlabel('PC 1', fontsize=12)
plt.ylabel('PC 2', fontsize=12)
plt.show()
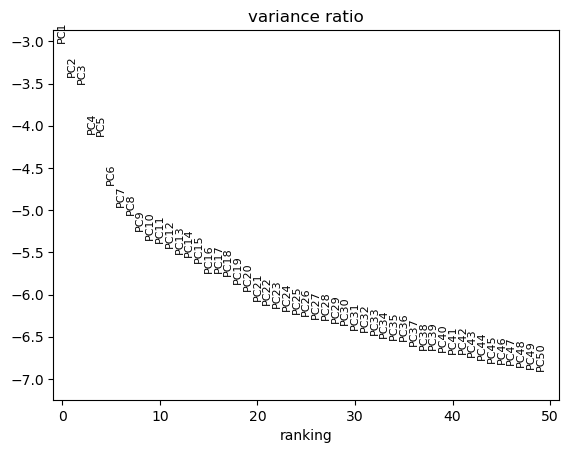
主成分贡献度:
n_pcs=50指定pc个数
# 主成分贡献度:
sc.pl.pca_variance_ratio(adata, log=True, n_pcs=50)
umap图:
# umap图
sc.pl.umap(adata, color=["leiden"], legend_loc="on data", size=5)
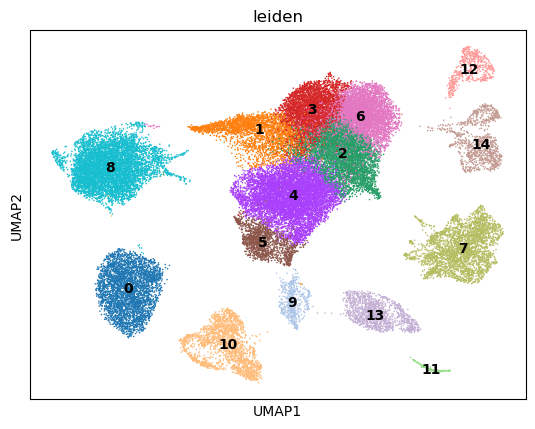
提取umap坐标:
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
umap_df = pd.DataFrame({
'UMAP_1': adata.obsm['X_umap'][:, 0],
'UMAP_2': adata.obsm['X_umap'][:, 1],
'Cluster': adata.obs['leiden']
})
umap_df.head()
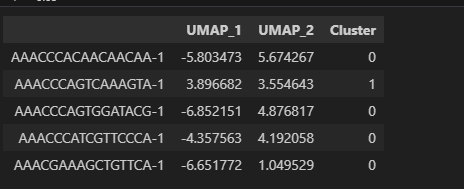
保存umap坐标:
# 保存为CSV文件
umap_df.to_csv('umap_df.csv', index=True) # index=True表示保存索引
美化 umap:
plt.figure(figsize=(5, 5))
# 使用 seaborn 绘制散点图,按 Cluster 着色
sns.scatterplot(
data=umap_df,
x='UMAP_1',
y='UMAP_2',
hue='Cluster',
palette='Set1',
alpha=0.5,
size=0.1,
legend=None# 不显示默认图例
)
# 计算每个 Cluster 的中心点并添加标签
unique_labels = umap_df['Cluster'].unique()
for label in unique_labels:
label_coords = umap_df[umap_df['Cluster'] == label]
center_x = label_coords['UMAP_1'].mean()
center_y = label_coords['UMAP_2'].mean()
plt.text(center_x, center_y, label, fontsize=12, ha='center', va='center')
# 图片设置
plt.grid(False)
plt.title('UMAP Plot with Cluster Labels', fontsize=14)
plt.xlabel('UMAP 1', fontsize=12)
plt.ylabel('UMAP 2', fontsize=12)
plt.show()
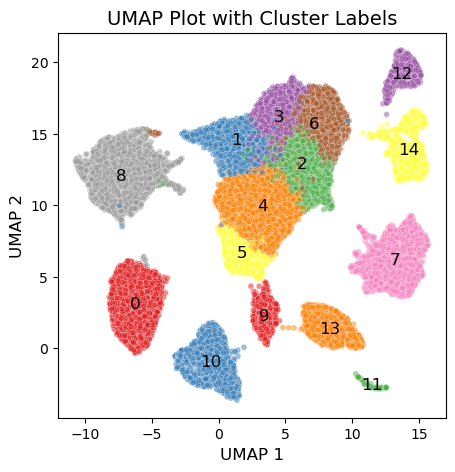
4.数据保存
保存 整个对象:
adata.write("GSE163558.adata.h5ad")
保存每一个细胞对应的信息,即metadata:
adata.obs.to_csv('GSE163558.adata.obs.csv', index=True)
提取子集:
adata_subset = adata[adata.obs['leiden'].isin(['0', '1'])].copy()
adata_subset
你学会了吗~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-04-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号