基于多序列比对结果的一致性序列鉴定
原创

1、概念
基于多序列比对(Multiple Sequence Alignment, MSA)结果的一致性序列鉴定是生物信息学中的一项重要任务,它帮助我们理解不同序列之间的保守性和进化关系。一致性序列(Consensus sequence)是指在多个相关序列的比对中,每个位置上出现频率最高的碱基或氨基酸组成的序列。
2、实现思路
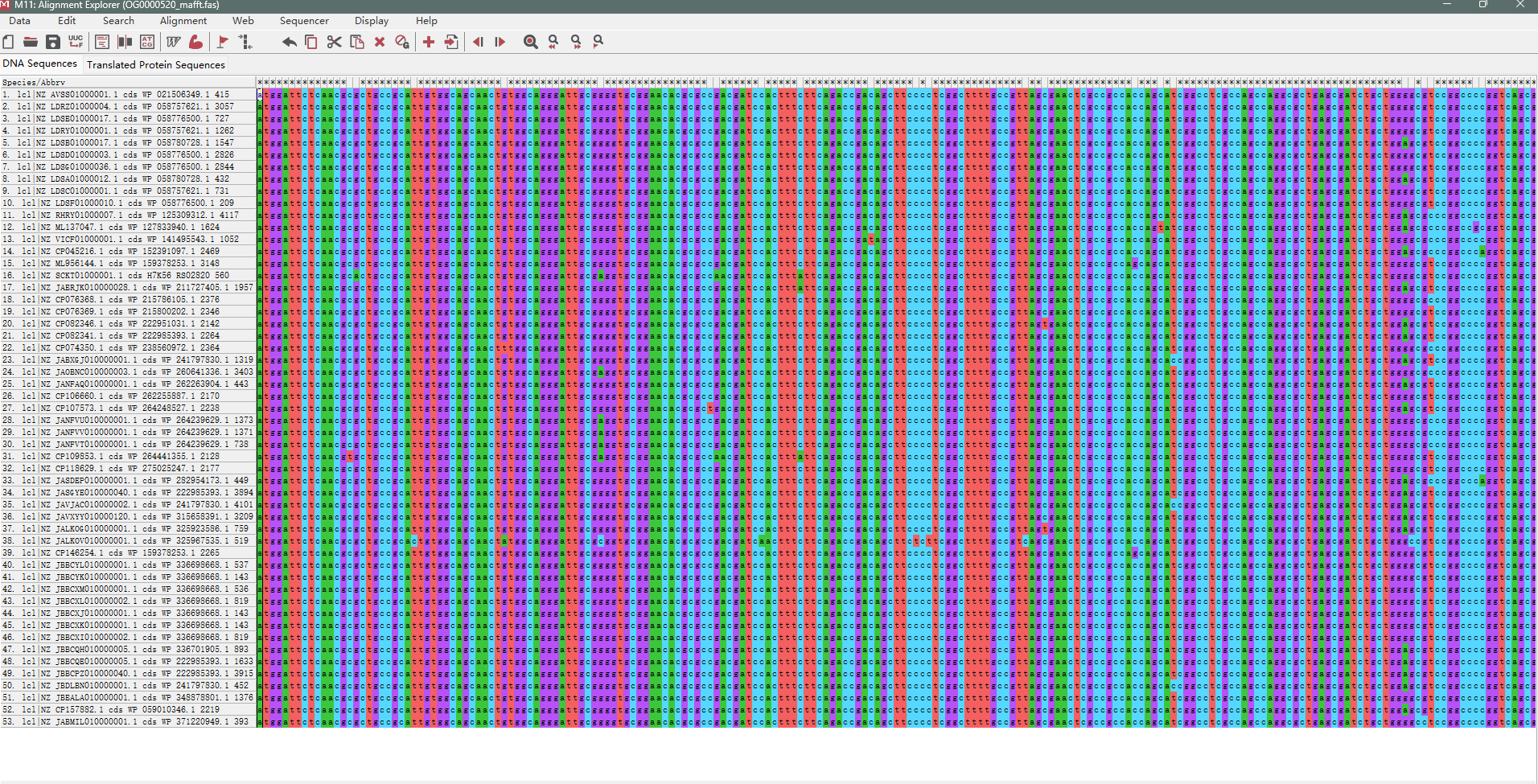
原文件用MEGA打开截图(部分)
我现在有多序列比对的结果文件,里面的内容如图所示(奇数行为序列的名字,偶数行为序列的内容),我现在需要做基于多序列比对结果的一致性序列鉴定,我的思路是每一个序列纵向比对,如果全都是一样的字母,那么则说明这个纵向一致,如果这个纵向序列不全一样的化,则说明这个纵向没有一致,如果有连续超过100个字符的纵向一致列存在,那么打印出来所对应的这样的每一小段序列,这就是我想要的一致性序列,最终输出在一个文件里。

多序列比对原文件
3、实现代码
from Bio import AlignIO # 导入Biopython中的AlignIO模块用于读取比对文件
import sys # 导入sys模块用于处理命令行参数
def find_consensus_regions(alignment, min_length=100):
"""
查找一致序列区域。
参数:
alignment (MultipleSeqAlignment): 多序列比对对象
min_length (int): 最小的一致性区域长度,默认为10个碱基
返回:
list of tuples: 包含开始和结束位置的一致性区域列表
"""
consensus_regions = [] # 存储找到的一致性区域
current_region_start = None # 当前一致性区域的起始位置
current_stretch = 0 # 当前连续相同字符的数量
# 遍历比对中的每一列
for i in range(alignment.get_alignment_length()):
column = alignment[:, i] # 获取当前列的所有序列字符
if all(base == column[0] for base in column): # 检查该列中所有字符是否相同
if current_region_start is None:
current_region_start = i # 如果没有开始位置,则设置当前列为起始位置
current_stretch += 1 # 增加当前连续相同字符的数量
else:
if current_stretch >= min_length:
# 如果当前连续相同字符数量达到或超过最小长度,则保存该区域
consensus_regions.append((current_region_start, i - 1))
current_region_start = None # 重置起始位置
current_stretch = 0 # 重置连续相同字符数量
# 检查最后一段连续相同字符是否满足条件
if current_stretch >= min_length:
consensus_regions.append((current_region_start, i))
return consensus_regions
def main():
"""
主函数,用于处理命令行参数并调用查找一致序列区域的函数。
"""
if len(sys.argv) != 2:
print("用法: python script.py <alignment_file>")
sys.exit(1)
alignment_file = sys.argv[1] # 获取命令行提供的比对文件路径
alignment = AlignIO.read(alignment_file, "fasta") # 读取FASTA格式的多序列比对文件
consensus_regions = find_consensus_regions(alignment) # 查找一致序列区域
with open('consensus_sequences.fasta', 'w') as output_file:
for start, end in consensus_regions:
# 提取一致性区域的序列,并写入输出文件
sequence = alignment[:, start:end+1]._records[0].seq
output_file.write(f">Consensus_{start}_{end}\n{sequence}\n")
if __name__ == "__main__":
main() # 如果脚本作为主程序运行,则调用main函数4、实现过程
1、将find_consensus.py和OG0000520_mafft.fas(多序列比对原文件)放在工作目录下
2、输入指令
输入指令
3、输出结果
生成consensus_sequences.fas文件在工作目录下
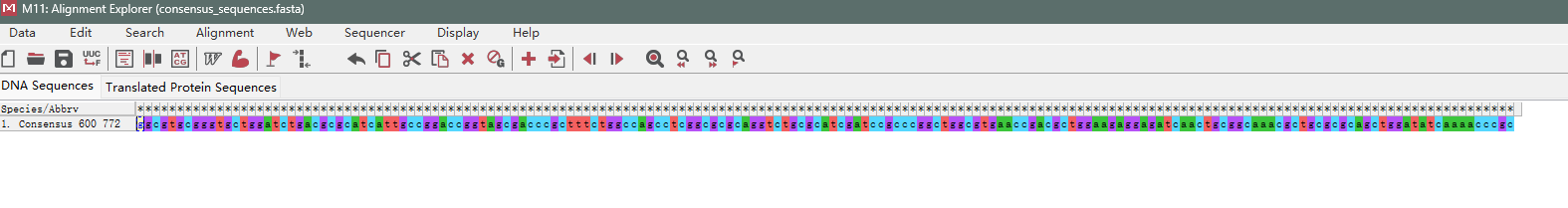
生成文件consensus_sequences.fas(全部结果)
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号