CAR-T治疗急性髓细胞性白血病的scRNA-Seq图谱


写在前面
这是一篇粉丝来稿(欢迎来稿,提供稿费的哦:少走二十年弯路),甚至贴心的为大家整理好了PPT(下载链接: https://pan.baidu.com/s/16-PGGGOVrAYcLCCl03BN9g?pwd=srpu)
嵌合抗原受体T细胞(CAR-T细胞)已经成为B细胞恶性肿瘤患者的一种强有力的治疗选择,但由于缺乏安全的治疗靶点,在治疗急性髓系白血病(AML)方面尚未取得成功。
做的事情:在这里,我们利用了一个公开可用的rna测序数据图谱,这些数据来自15名AML患者和9名健康患者的超过50万个单细胞,用于预测恶性细胞中表达但健康细胞中缺乏的目标抗原,包括T细胞。在这种高分辨率的单细胞表达方法的帮助下,我们计算确定了集落刺激因子1受体和簇分化86作为CAR-T细胞治疗AML的靶点。这些建立的CAR-T细胞的功能验证表明,在细胞系和人类衍生的AML模型中,CAR-T细胞在体外和体内都具有强大的有效性,对相关健康人体组织的脱靶毒性最小。
然而,靶向非b细胞相关表位的CAR-T细胞尚未显示出类似的反应率。例如,在髓系恶性肿瘤中,如急性髓系白血病(AML),常见的靶结构通常在重要组织中共同表达,如内皮细胞或造血干细胞和祖细胞(hspc),增加了靶向非肿瘤毒性的风险6,7。因此,确定安全的靶结构对于CAR-T细胞治疗髓系肿瘤的巨大潜力至关重要。AML是成人中最常见的急性白血病,其分子异质性使新治疗药物的成功开发变得复杂8。尽管大多数采用联合化疗的患者有早期治疗的意图,但疾病复发仍很频繁,超过50%的患者会出现复发9。复发后,异基因造血干细胞移植(allogenehematopoieiestem cell transplantation, allohsct)仍然是唯一的治疗方法;但即便如此,长期生存的可能性仍低于20%(长期的生存可能性低)。因此,创新的治疗方案代表了很高的未得到满足的医疗需求。
现阶段针对AML的治疗的靶向研究进展:目前,靶向aml相关靶抗原CD33和白细胞介素-3受体-α (IL3RA, CD123)的CAR-T细胞正在进行临床研究。(但是依然存在着脱靶的现象)由于临床前证据表明对hscs具有非肿瘤毒性,大多数临床试验都在评估抗cd123或抗cd33 CAR-T细胞作为异源hsct前移植过渡方案的潜力。这些试验的早期报告显示只有有限的治疗效果10 - 12。然而,这些AML临床研究的更完整的结果仍在等待中。同时,其他靶点,如CD70、c型凝集素样分子-1、fms样酪氨酸激酶-3 (FLT3)、CD44变异体6 (CD44v6)、唾液酸结合igg样凝集素-6 (sigec -6)或CD117,已在临床前研究中作为替代CAR靶点进行了测试13 - 17。然而,临床验证尚未完成,大多数靶点的表达谱至少增加了一些关于其临床安全性和有效性的不确定性。(寻找新的靶标的必要性)
新开发的CAR-T细胞通常定向于已经用于抗体治疗的目标结构。相比之下,对CAR-T细胞治疗进行无偏的从头靶向筛选却很少进行。此外,直到最近,非肿瘤抗原投影只能利用大量测序数据,缺少关于细胞类型特异性靶抗原表达模式的详细信息。很方便的是,过去十年单细胞技术的革命产生了大量的单细胞表达数据集,这些数据集提供了健康和恶性细胞的精确的转录组解剖信息19,这是一个几乎未被开发的治疗开发资源,至少在从头抗原预测和CAR-T细胞发展的背景下。这些进展允许深入的肿瘤和非肿瘤抗原预测,以无与伦比的分辨率为健康和恶性细胞提供了独特的见解。
主要结果
结果1 基于scrna序列的筛选算法开发
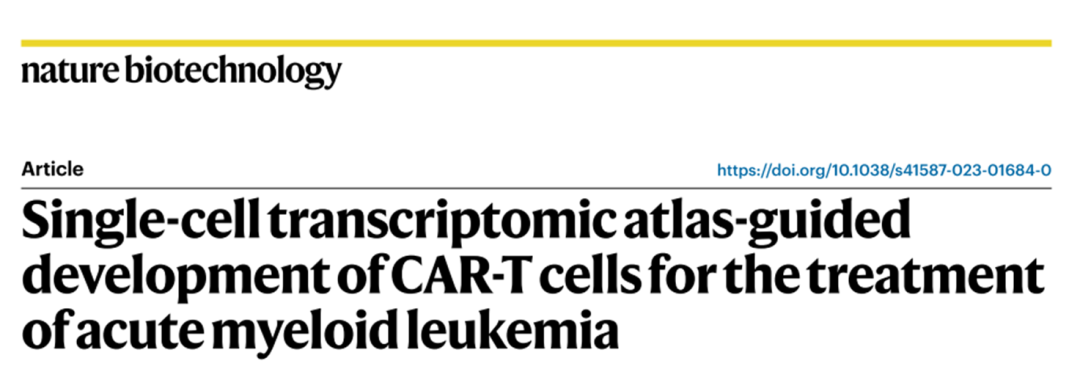
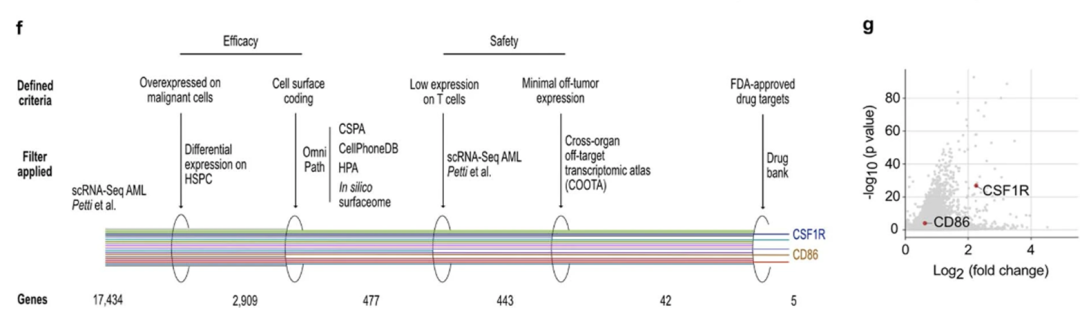
我们创建了一种基于scrna序列的无偏发现方法来识别CAR目标。为了确保CAR的有效性,一个合适的候选基因是(1)在恶性细胞中过表达(2)位于细胞表面。在CAR安全性方面,候选CAR应该(3)在T细胞上不表达,(4)在重要的健康组织中显示最小的表达(图1a)。
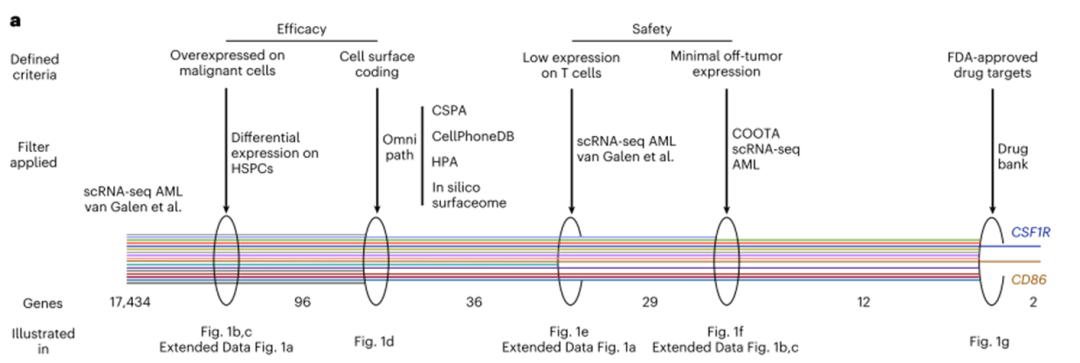
将我们的方法应用于AML,我们使用了来自15名AML21患者的公开可用的scRNA-seq数据。从这些样本中,共有28,404个经测序的健康和恶性骨髓细胞通过了质量控制(图1b,c;有关质量控制步骤的详细描述,请参阅方法)。
为了获得最大的CAR疗效,我们试图确定在恶性hspc样细胞(此处称为造血干细胞(HSC)样和祖细胞(Prog)样)上比在健康细胞上表达更高的候选细胞。恶性和健康hspc之间的差异基因表达分析显示,有96个基因在hspc样细胞中强烈过表达,并被用于进一步的下游分析(扩展数据图1a)。
为了确定CAR-T细胞在靶细胞表面可获得的候选基因,我们使用了OmniPath22,一个大规模的分子数据库,将来自多个资源的数据整合到一个包含4924个基因的综合人类表面基因库中(图1d)。
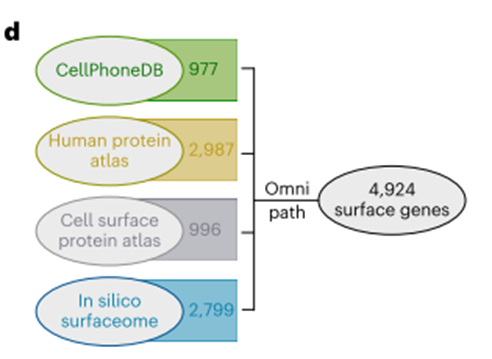
在96个hspc样细胞中过表达的基因中,有36个出现在这个文库中。通过之前所有过滤但在T细胞上表现出高表达的基因(例如CD52和CRIP1)被排除在进一步的分析之外(图1e)。

为了减少靶内非肿瘤效应,我们处理和协调了来自9个健康人体组织(大脑、肺、淋巴结、心脏、皮肤、肝脏、肾脏、结肠和食道)的11个scRNA-seq数据集,形成一个由超过500,000个单一健康细胞组成的大规模跨器官非靶转录组图谱(COOTA)(图1f) 。在扩展数据图1b,c中提供了用于COOTA的所有数据集的详细总结。在重要的非免疫细胞谱系或直接接近注入T细胞的细胞类型组织(即内皮细胞、动脉、静脉、支气管血管、毛细血管和平滑肌细胞)中高度表达的靶细胞被排除在进一步的分析之外(图1f)。
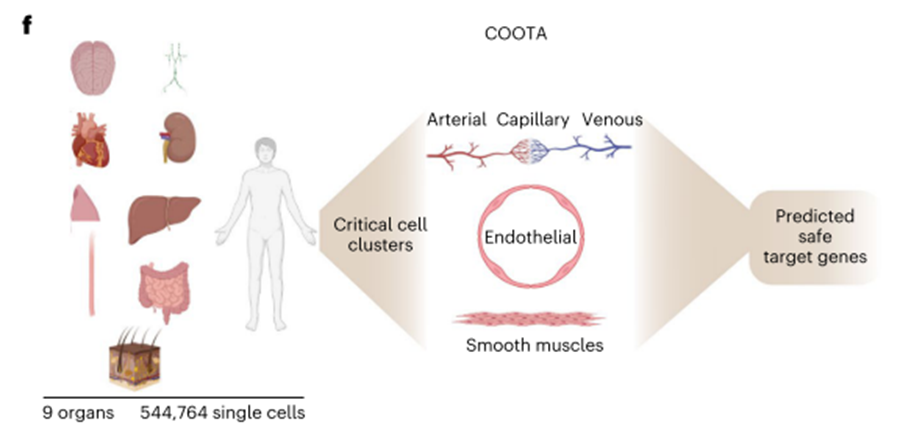
使用这种严格的方法,CAR开发仍然有12个潜在的候选者。有趣的是,大多数描述的AML CAR靶点(n = 20)在不同水平上未能达到我们分析的阈值(扩展数据图1d)。
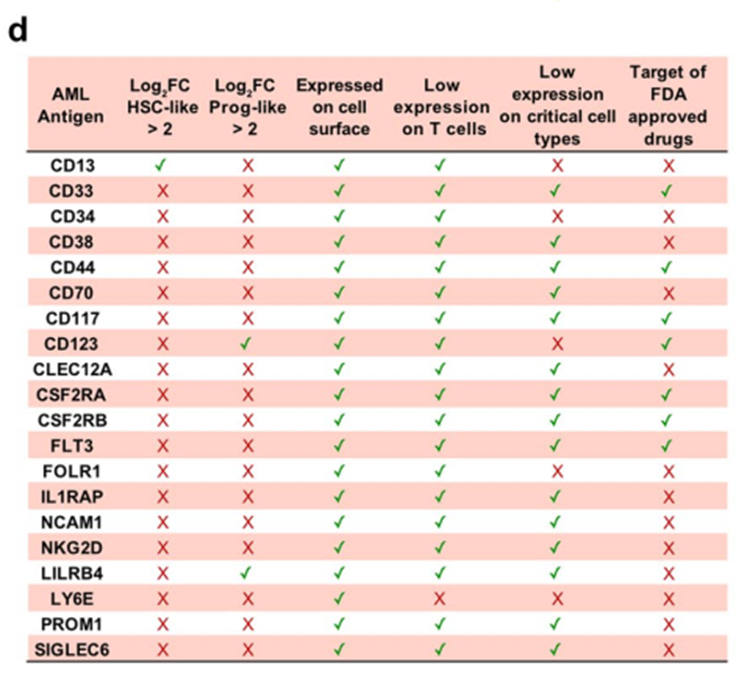
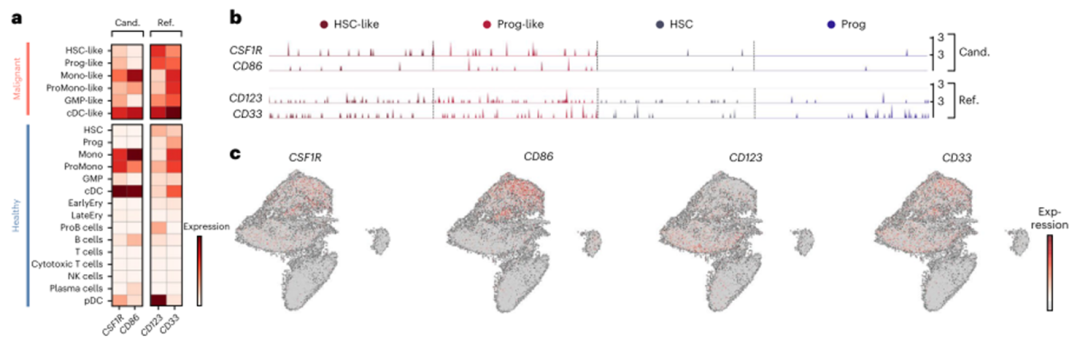
例如,原型AML抗原CD33和CD123不符合我们在恶性热休克细胞中过表达的严格标准(见应用阈值方法),最有可能是由于这两种抗原在健康热休克细胞中表达。此外,CD123在内皮细胞和各种肺细胞类型中均有高表达(具体分析见图2d)。(同时也通过其他的靶点侧面证实了这种分析方法的有效性)
为了进一步优化新开发的CAR-T细胞的安全性,我们推断,如果针对12个候选细胞中的任何一个的靶向治疗已经被食品和药物管理局(FDA)批准,新开发的CAR-T细胞发生意想不到的、严重的靶向非肿瘤毒性的风险将被最小化。此外,这可以缩短时间长度,并减少将新开发的CAR-T细胞转化为临床常规的调节障碍,因为靶向治疗的安全性已经被证明。因此,我们使用了一个可访问的数据库,其中包含了所有被监控的fda批准的药物,其中包含了药物和药物靶标的相互作用、药理学和化学结构的信息38。我们确定了两个靶点,CD86和CSF1R,它们已经进行了临床研究(图1g)。据我们所知,抗cd86和抗csf1r CAR-T细胞都没有被用于AML的CAR-T细胞治疗。因此,我们决定进一步研究它们的潜力。
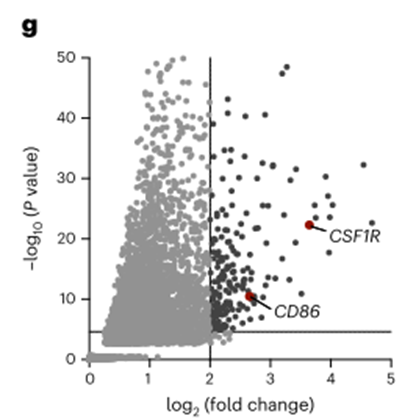
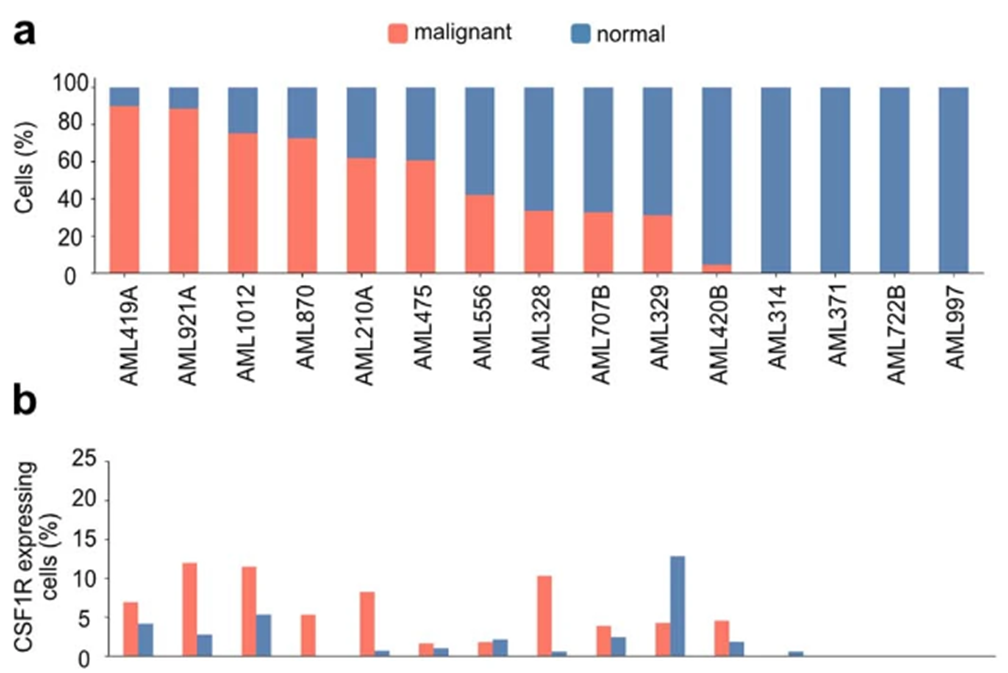
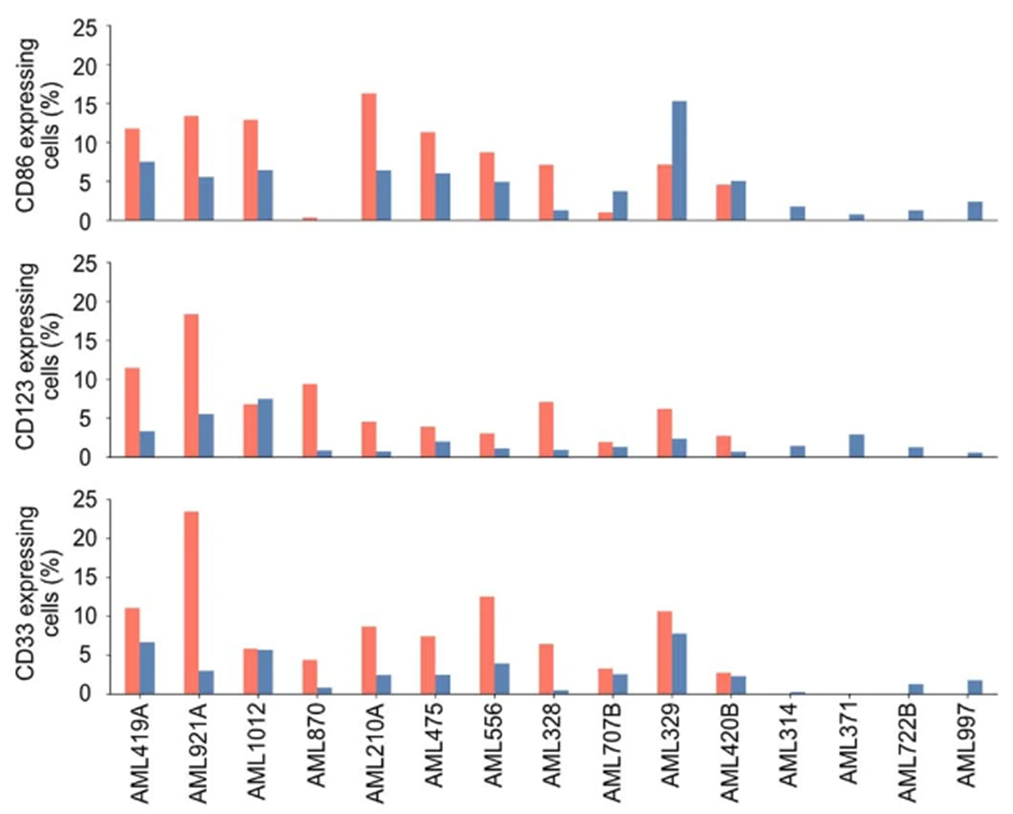
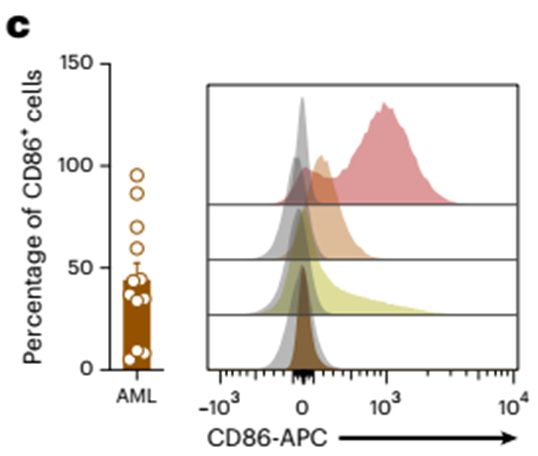
在捕获恶性母细胞的AML患者中,这两种抗原在恶性细胞间高度表达(15人中有11人;图2a,b),尽管参与者群体的分子分布不均匀。
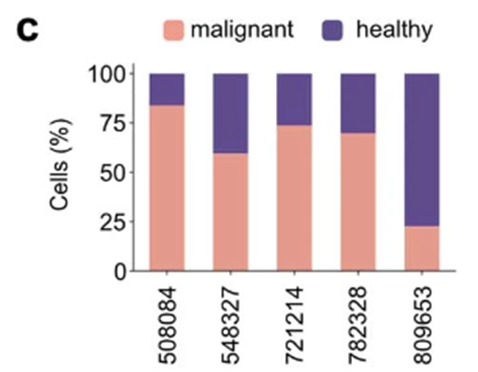
为了确保我们分析的有效性,并更好地反映AML作为一种疾病的细胞遗传学多样性,我们下一步试图进一步增加队列的规模。因此,我们获得了第二个公开可用的scRNA-seq数据集,其中包含另外5个具有AML39的个体(扩展数据图2c)。
为了交叉验证我们的计算目标识别方法,我们使用了scANVI,一种半监督变分autoencoder,将Petti等人生成的参考图映射到一个新的参考图中(扩展数据图2e)。
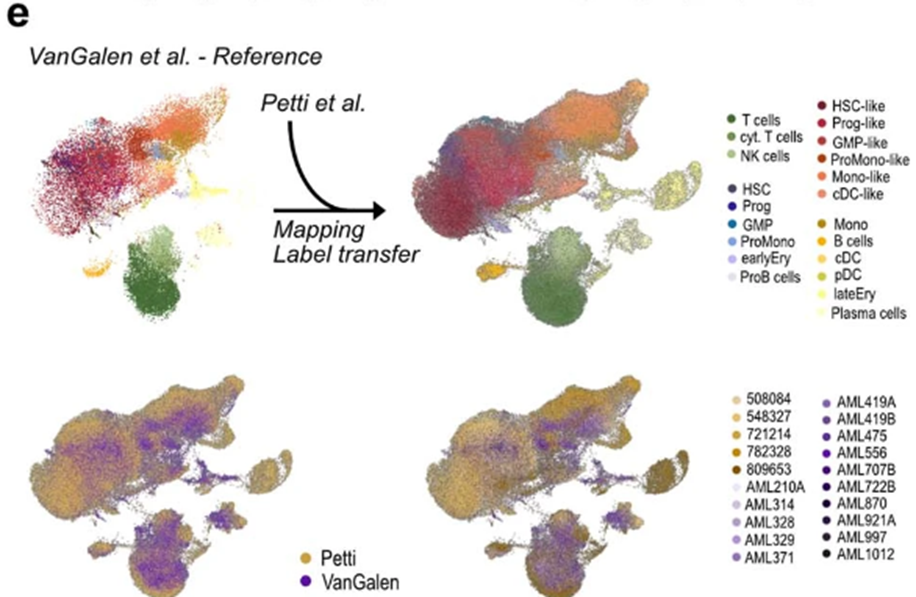
综上所述,CSF1R和CD86在恶性造血细胞中比在健康造血细胞中优先表达。接下来,在将我们的目标识别方法扩展到这5个额外的AML患者后(图1a), CSF1R和CD86在第二个AML队列中再次被确定为CAR治疗的合适目标抗原(扩展数据图2f,g)。
总之,使用两个独立的单细胞AML队列,共包括20个个体,我们确定CSF1R和CD86作为AML治疗的潜在CAR靶点。
结果2:CSF1R和CD86在肿瘤发生和消失时的表达分析
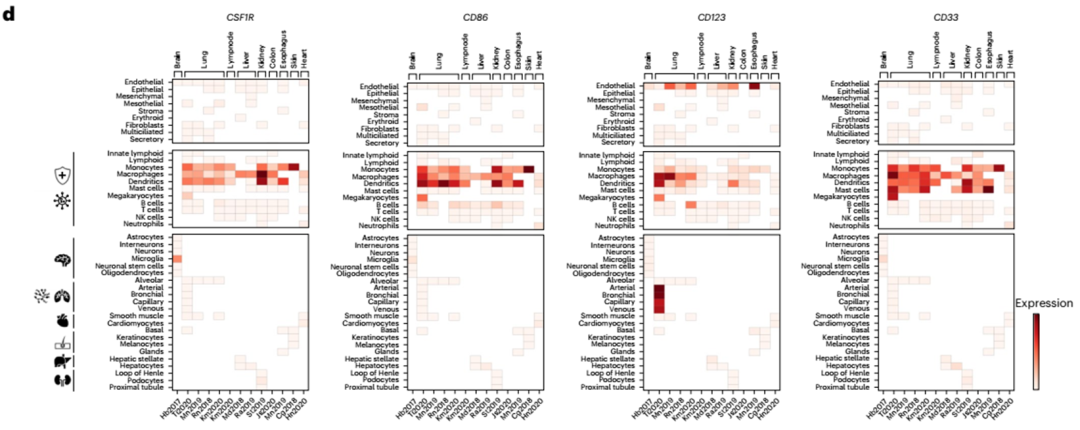
接下来,我们将两个靶抗原CSF1R和CD86与参考基因CD123和CD33进行比对,以便在转录组水平上更容易解释受体的表达(图2a-c)。CSF1R在所有6个恶性细胞簇中均有表达,但在单核细胞样或常规树突状细胞样簇中表达最高。CD86在单核细胞样、前细胞样和常规树突状细胞样簇中表达最强(图2a)。在恶性HSPC簇中,CSF1R的表达高于CD86,但低于CD123和CD33内参基因(图2a,b)。相反,在健康的造血干细胞和祖细胞中检测到CD123或CD33,而CSF1R和CD86仅在这些细胞中表达极少(图2b)。通过统一流形近似和投影(UMAP)嵌入可见,CSF1R和CD86的表达谱与CD123和CD33内参基因非常相似(图2c)。
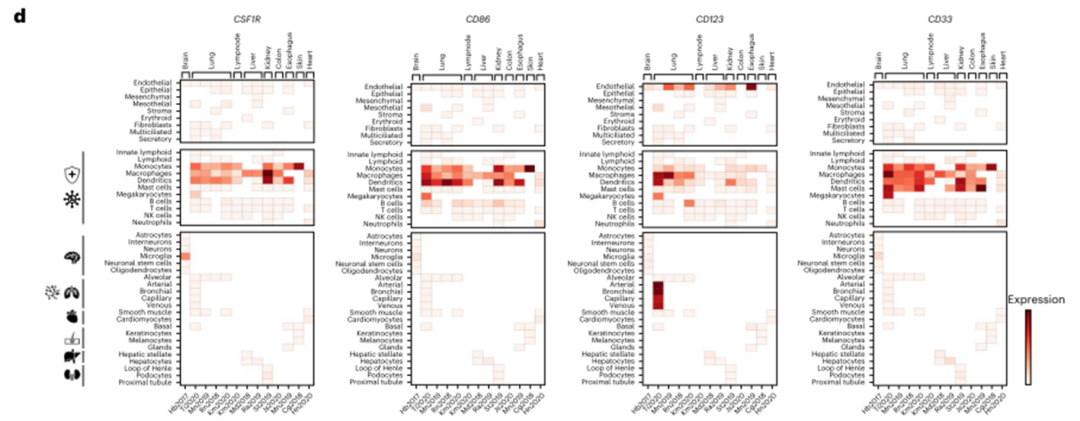
COOTA分析显示,靶抗原主要在骨髓来源的免疫细胞(单核细胞、巨噬细胞和树突状细胞)中表达,与CD33的外周表达谱相似(图2d)。CSF1R和CD86在上皮细胞或基质细胞上没有高表达(图2d,上)。在器官特异性细胞簇中(图2d,底部),表达仅限于大脑中的小胶质细胞,如文献所述。
接下来,我们试图在蛋白质水平上评估目标抗原的表达。我们使用六种不同的人AML细胞系(THP-1、Mv4-11、OCI-AML-3、PL-21、MOLM-13和U937)和B细胞恶性NALM-6细胞作为阴性对照细胞进行了初步筛选(图2e)。所有筛选的AML细胞系均检测到CSF1R和CD86。检测CD123和CD33的表达作为参考(图2e)。考虑到我们目标的成熟、健康的免疫细胞对CD33的表达类似,我们决定使用CD33作为后续所有实验的主要对照。
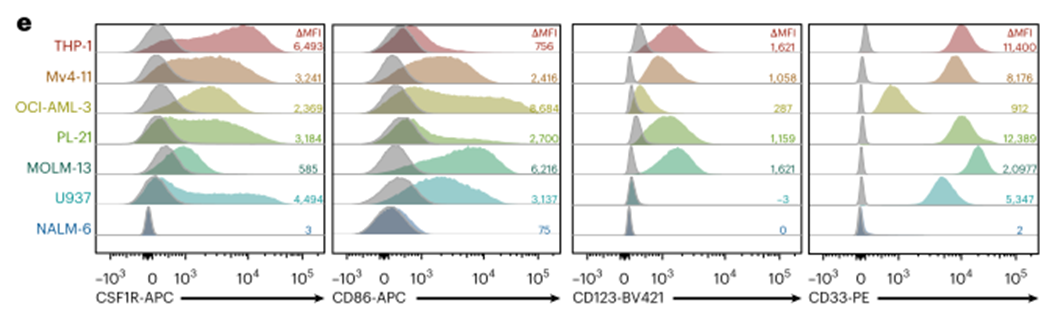
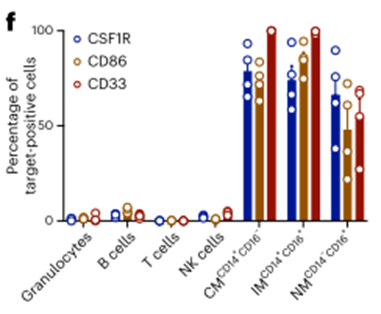
为了验证COOTA预测的转录组谱,我们使用多色流式细胞术评估健康供者外周血免疫细胞中每个候选抗原的受体表达(图2f)。根据我们的转录组预测,CSF1R和CD86的表达主要局限于单核细胞群体,在粒细胞或T细胞上没有表达(图2f)。
结果3 抗小鼠CSF 1 R CAR-T细胞(mCSF 1 R CART)不会在小鼠中引起毒性
尽管我们的方法和深入的非肿瘤抗原投射设定了严格的阈值,CSF1R和CD86的表达模式仍然比临床使用的候选者(CD19和BCMA)更广泛,这些候选者几乎完全限于B细胞或B细胞亚群11。因此,我们首先在具有完全免疫能力的同基因小鼠模型中测试了开发的抗靶向CAR-T细胞的安全性。为了确保小鼠和人类的靶基因表达相似,我们利用现有的批量测序数据,比较了不同器官中候选基因的表达(图3a)。
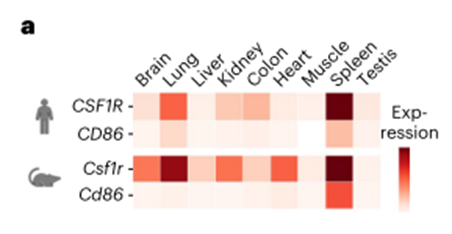
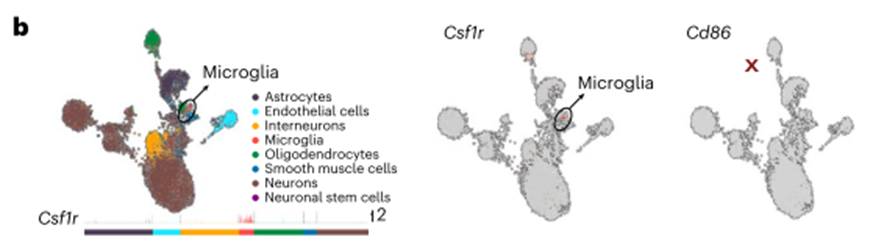
CSF1R在小鼠和人的器官中均有较高的表达,而CD86仅在脾脏中检测到。此外,与我们的COOTA预测一致,CSF1R已知在微胶质细胞42上表达,这增加了额外的安全问题。对存档的小鼠脑组织进行的scRNA-seq分析证实了Csf1r在小胶质细胞中表达,在组织驻留的骨髓细胞中也有类似的表达模式(图3b)。
综上所述,我们决定使用CSF1R来建立潜在的小鼠脱靶毒性模型。我们对产生mCSF1R抗体的杂交瘤进行了测序,并设计了第二代mCSF1R CART(扩展数据图3a)。小鼠抗epcam CAR-T细胞(mEpCAM CART)或mcherry转导的T细胞作为所有实验的阴性对照(扩展数据图3a)。mCSF1R CAR构建物可以有效地转导到小鼠原代T细胞中(图3c)。通过fc固定的重组小鼠CSF1R蛋白剂量依赖性地激活mCSF1R CART,与mEpCAM CART相比,激活标记物CD69(图3d,左)和脱颗粒标记物CD107a细胞表面暴露(图3d,右)可见。
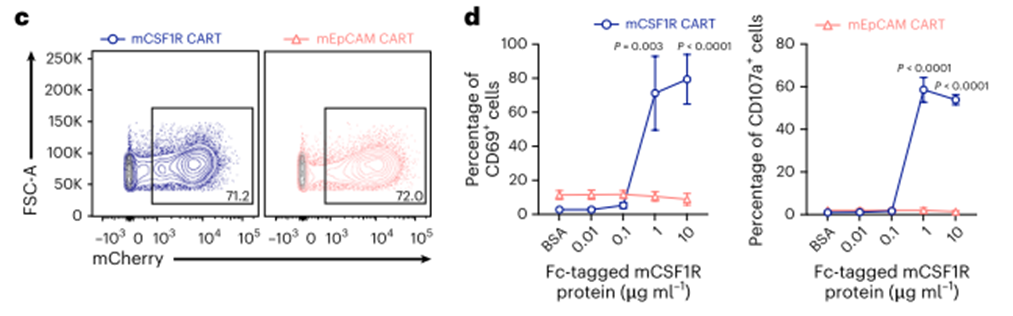
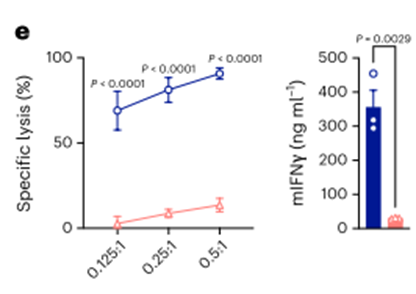
为了进一步验证开发的mCSF1R CART的功能,我们研究了对表达mCSF1R的细胞系的杀伤能力。因此,我们选择小鼠网状细胞肉瘤细胞株J774A。1,表达mCSF1R43。利用流式细胞术,我们验证了mCSF1R在J774A上的表达。而mEpCAM未检测到(扩展数据图3b)。mCSF1R或mEpCAM CART与J774A共培养。1例肿瘤细胞显示J774A的高效裂解。用mCSF1R CART检测1个肿瘤细胞(图3e,左)。作为选择性激活的标志,mCSF1R CART分泌大量的干扰素-γ (IFNγ)(图3e,右)。
接下来,我们使用体内实验来评估靶内毒性的风险。最初,mCSF1R CART或对照组经静脉(静脉)注射到健康的C57BL/6小鼠中进行有限的植入(扩展数据图3c-e)。
为了增强T细胞的持久性,小鼠接着进行全身照射预处理(WBI;5 Gy) mCSF1R CART过继细胞移植(ACT)前5 d(图3f)。高计数的mEpCAM CART作为阳性对照,而mCherry T细胞作为阴性对照。。在T细胞转移后,我们没有检测到mCSF1R cart处理小鼠作为小鼠毒性敏感替代物的体重变化(图3g)。相比之下,如文献44所述,经mEpCAM cart处理的小鼠在ACT后1周体重迅速下降(图3g)。
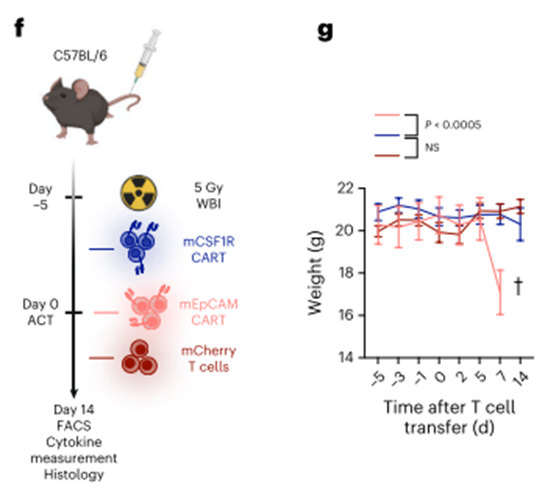
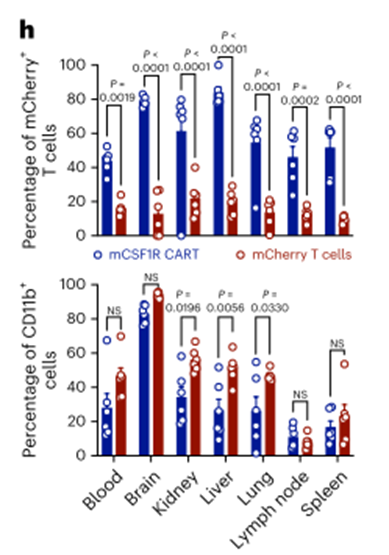
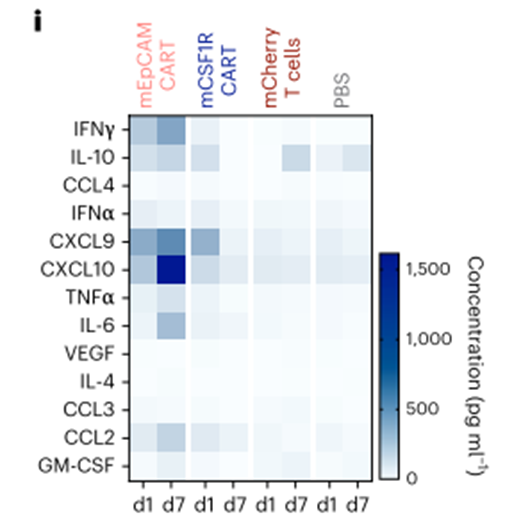
第7天,当mEpCAM cart处理的小鼠达到预定的实验终点标准时,收集器官进行后续分析。剩余的mCSF1R CART-或mCherry T细胞处理的小鼠在ACT后2周被杀死,用流式细胞术分析器官来源的细胞悬液。我们在所有器官中检测到的mCSF1R CART细胞的比例高于经mCSF1R转导的T细胞,这表明mCSF1R CART细胞比经mCSF1R转导的T细胞具有更好的持久性(或抗原依赖性增殖)(图3h,顶部。我们观察到在肾、肝和肺中组织驻留的CD11b+细胞数量减少,但在其他分析器官中没有(图3h,底部),这很可能是由于mCSF1R CART的靶向作用。ACT后第1天或第7天的多重血清细胞因子测量显示,mCSF1R CART和mCherry T细胞或pbs处理的小鼠在第7天的细胞因子水平没有差异(图3i)。相比之下,接受mEpCAM CART的小鼠血清中检测到高水平的促炎细胞因子,如IFNγ、CXCL9或CXCL10(图3i)。
类似地,mEpCAM CART治疗的小鼠血清中临床使用的器官损伤标志物(如尿素、胆红素和肝酶)水平升高,而mCSF1R CART或mCherry T细胞治疗的小鼠则没有升高(扩展数据图3g)。最后,我们对已知CSF1R高表达的器官进行了组织病理学分析。mCSF1R car处理的小鼠在苏木精和伊红染色的肺、肝脏或脾脏中没有出现任何器官损伤的迹象(扩展数据图3h)。值得注意的是,正如之前报道的44,经mEpCAM CART处理的小鼠肺显示肺泡上皮增厚,表明转移的mEpCAM CART具有靶向非肿瘤毒性(扩展数据图3h)。
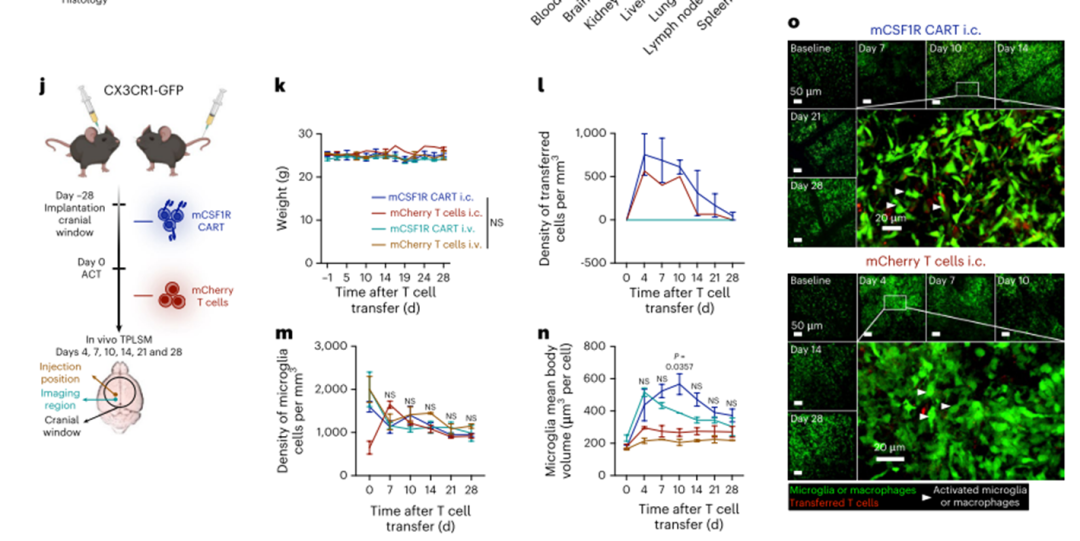
为了研究CAR-T细胞在大脑中的归位和杀伤潜力,我们使用了CX3CR1-GFP报告小鼠,通过双光子激光扫描显微镜(TPLSM)可以直接显示CAR-T细胞-小胶质细胞相互作用。颅窗植入后,将mCSF1R CART或mcherry转导的T细胞静脉或颅内(i.c)植入CX3CR1-GFP报告小鼠(图3j-o)。使用TPLSM监测T细胞-小胶质细胞相互作用、小胶质细胞形态的改变和小胶质细胞总数的减少,共28天(图3l-o)。我们再次观察到,在所有治疗组中,体重或行为均无变化(图3)。我们在i.c.植入mCSF1R CART或mCherry T细胞后检测到高T细胞数量(图3l,o)。无论小鼠是否植入mCSF1R CART或mCherry T细胞,这些数字在28天的疗程中逐渐下降。第28天,在任何组中均未检测到转移的T细胞(图3l,o)。此外,小胶质细胞的数量在任何组之间都没有本质上的差异(图3m,o)。i.c.植入T细胞后,小胶质细胞的平均体积增加,很可能是由于细胞的激活45(图3n,o)。这种激活在注射mCSF1R CART的小鼠中最为显著(图3n,o)。然而,到第28天,各组小胶质细胞活化的迹象减弱(图3n,o)。在静脉注射T细胞后,我们没有检测到mCSF1R CART和mCherry控制的T细胞在大脑中,也没有观察到小胶质细胞激活或耗尽的迹象(图3k-o和扩展数据图3i)。
我们的结果表明,尽管目标抗原在不同器官的组织驻留免疫细胞和小胶质细胞上表达,但没有相关的安全信号会阻止进一步的治疗发展。
结果4 抗人CAR-T细胞在AML异种移植模型中表现出高效能
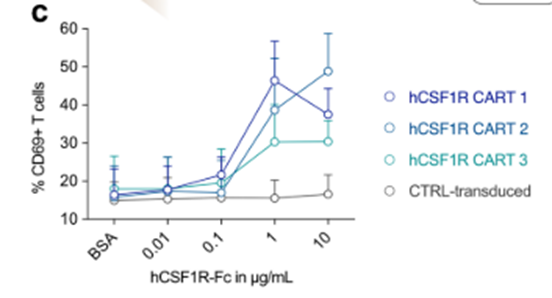
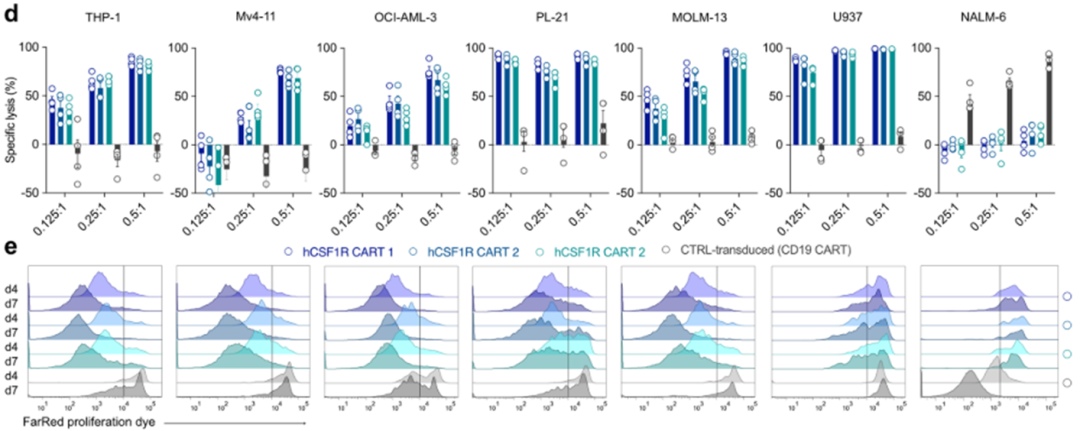
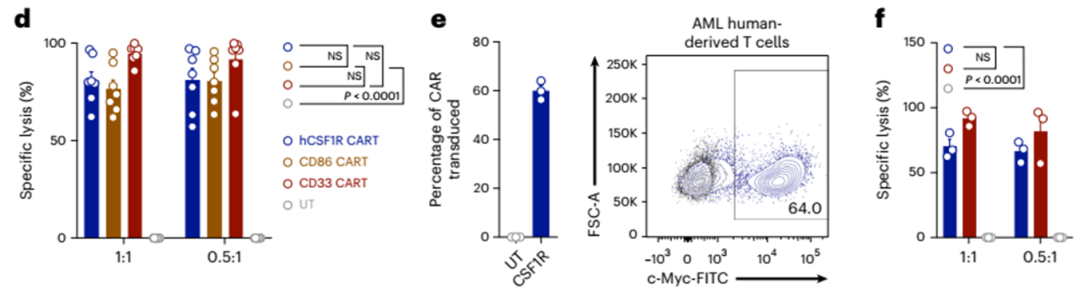
在证明CSF1R在各种同基因小鼠模型中的安全性后,我们下一步的目标是在人类模型中验证该靶点。我们克隆了一个抗人csf1r结合的单链片段变量(scFv)到现有的抗mcsf1r CAR主干中,这允许直接交叉比较在小鼠和人类中抗小鼠和抗人类CAR- t细胞的激活阈值。此外,我们还创建了两个包含CD8或CD28铰链结构域的完整的人抗csf1r CAR结构体(hCSF1R CART 1-3;扩展数据图4a(左)。首先,我们广泛交叉比较了不同anti-hCSF1R CAR结构的功能。所有构建物都可以有效地引入原代人T细胞(扩展数据图4b),并由重组板结合hCSF1R蛋白剂量依赖性地激活(扩展数据图4c)。CAR产品有效地裂解了所有6个经测试的人类AML细胞系,但不是抗原阴性的NALM-6细胞(扩展数据图4d)。CD8铰链结构域的构建在效靶细胞比例较低(E:T)的情况下显示出较高的溶解能力(扩展数据图4d)。为了评估抗原特异性增殖,我们将CSF1R CAR-T细胞与AML细胞共培养4或7天。所有的CSF1R CAR-T细胞均显示抗原特异性、时间依赖性增殖(扩展数据图4e)。T细胞数量的绝对定量显示,基于CD8铰链的抗csf1r CAR构建更加稳健的扩展(扩展数据图4f)。所有CSF1R CAR-T细胞在与THP-1、Mv4-11或OCI-AML-3 AML细胞共培养后分泌大量的IFNγ,但在与NALM-6对照细胞共培养时不分泌(扩展数据图4g)。基于这些结果,我们决定进一步研究含有CD8铰链结构域的CSF1R CAR-T细胞(hCSF1R CART 1,此处命名为hCSF1R CART)。
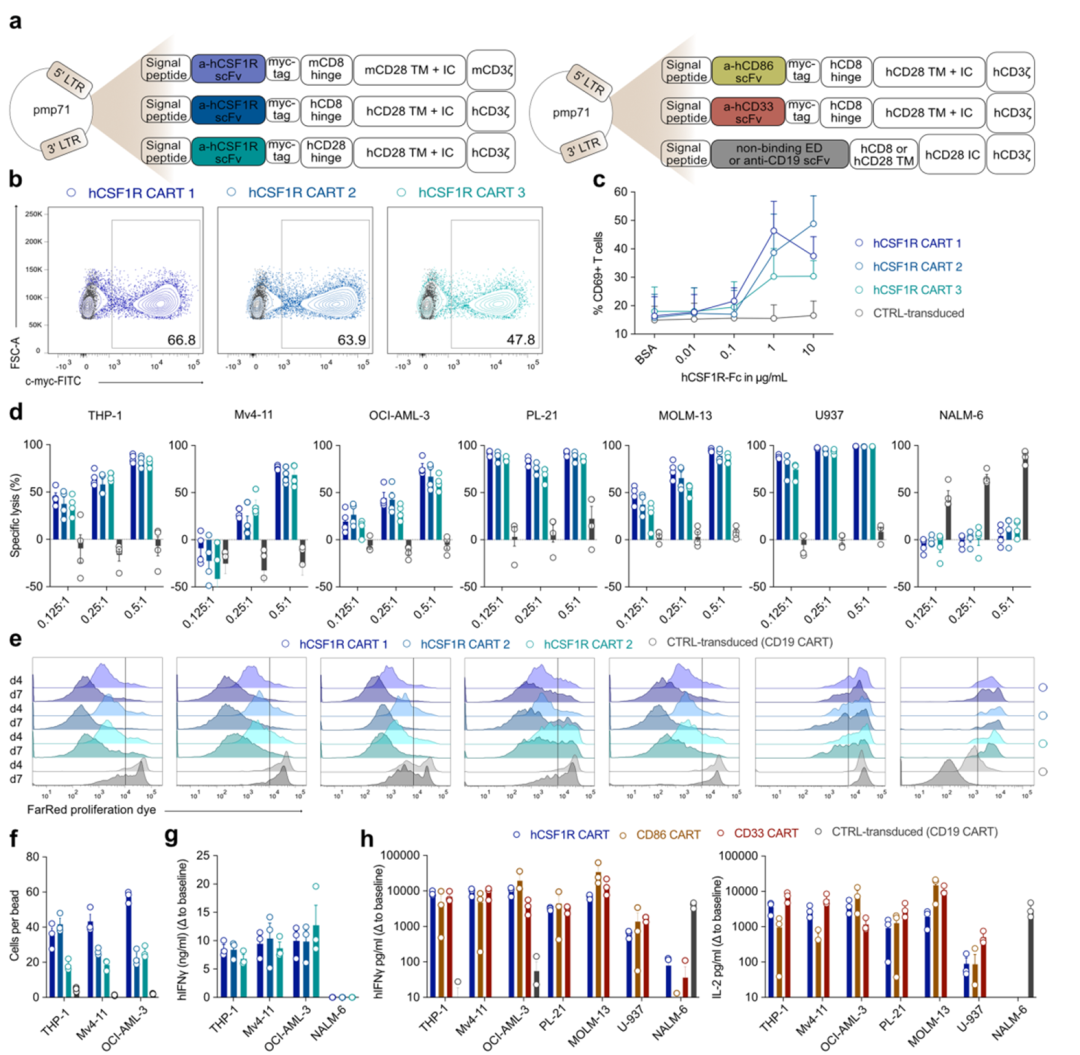
人类CD86 CAR-T细胞(CD86 CART)和人类CD33 CAR-T对照细胞(CD33 CART)的构建被类似地设计(扩展数据图4a,右)。所有CAR-T细胞产品都可以有效地导入原代人类T细胞(图4a)。为了验证CD86 CART的功能,并比较两种新开发的治疗方法的敏感性阈值,两种CAR-T细胞都与各自的平板结合抗原孵卵(图4b)。在极低浓度的靶蛋白(0.01µg ml-1)下,CD86 CART已经被激活。相比之下,hCSF1R CART需要1µg ml-1或更高的浓度(图4b)。我们将所有CAR-T细胞与AML细胞株共培养,并评估了AML细胞的特异性裂解和抗原依赖性增殖(图4c,d)。hCSF1R和CD86 CART有效地裂解了所有6个AML细胞系,与CD33 CART相当(图4c),并且增殖程度相似(图4d)。CD19 CAR-T细胞(CD19 CART)作为对照转导细胞。
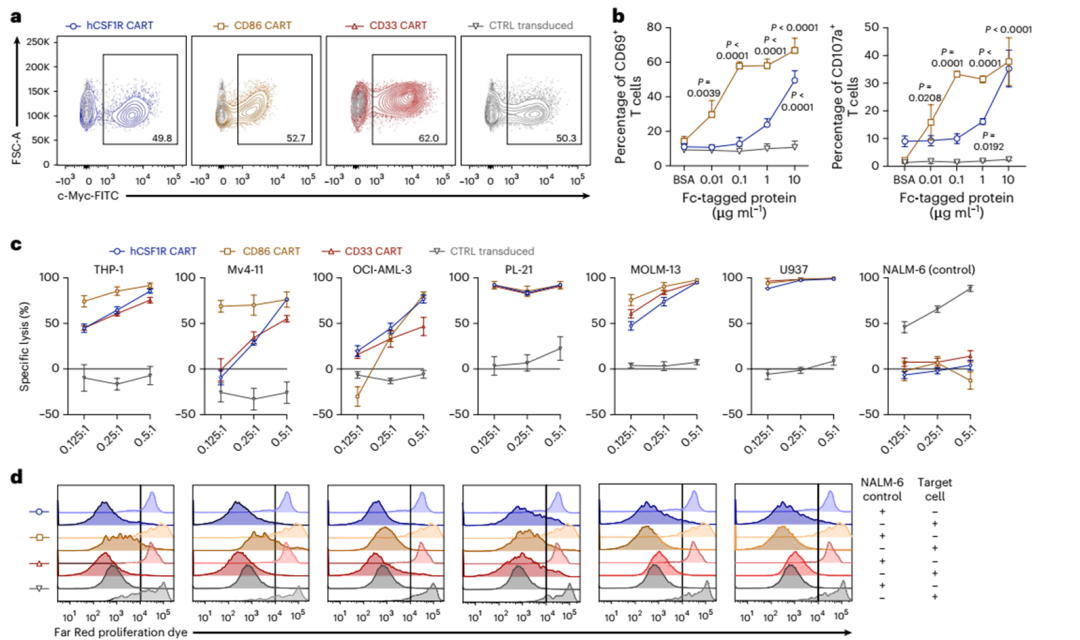
为了证明新开发的CAR-T细胞在体内的有效性,我们给NOD-scid Il2rgnull (NSG)小鼠注射致命剂量的Mv4-11 AML细胞,并用hCSF1R, CD86, CD33或CD19(对照)CAR-T细胞治疗(图4e)。我们使用生物发光成像(BLI)监测肿瘤进展。hCSF1R和CD86 CART均能在体内消除Mv4-11的肿瘤负荷(图4f-h)。为了给另一种AML模型提供体内证据,我们将致命剂量的THP-1细胞静脉注射到NSG小鼠体内,并用CSF1R、CD33或对照CART治疗它们(图4e)。再次,hCSF1R CART有效地控制了实验性白血病,具有与CD33 CART相似的完全缓解(CR)率(hCSF1R CART: CR在十分之七;CD33 CART: 8 / 10为CR),肿瘤细胞注射后总生存期达80 d(图4i-k)。总之,我们能够在体外和体内证明新开发的hCSF1R和CD86 CART对一大批人类AML细胞系的有效性。
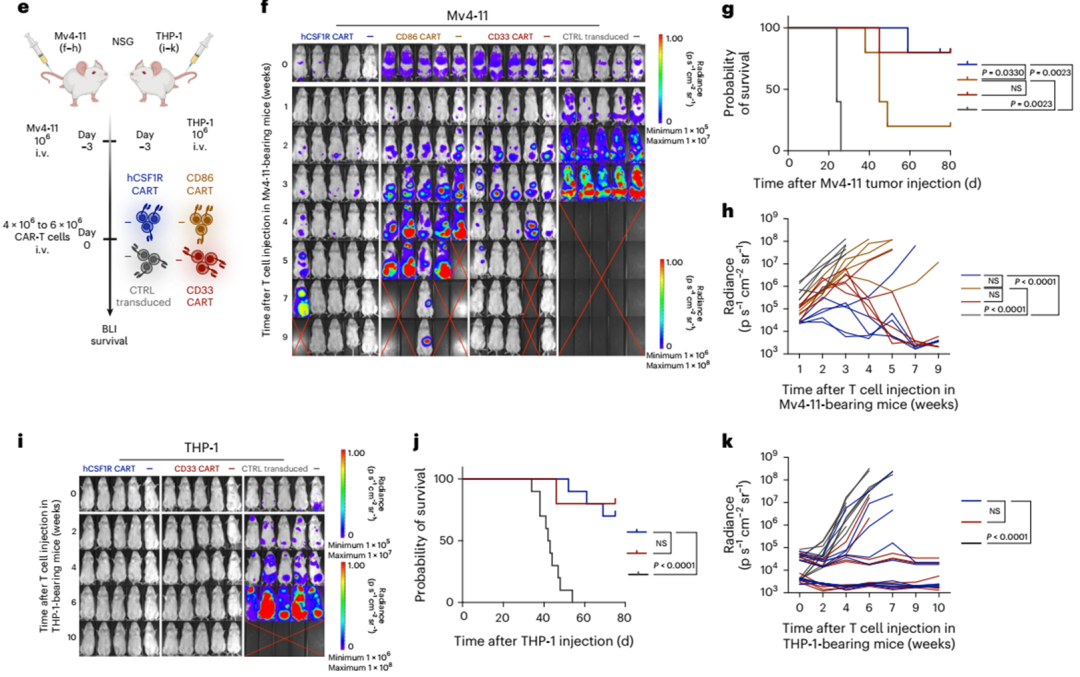
结果5 hCSF1R和CD86 CART在初级人类模型中是有效的
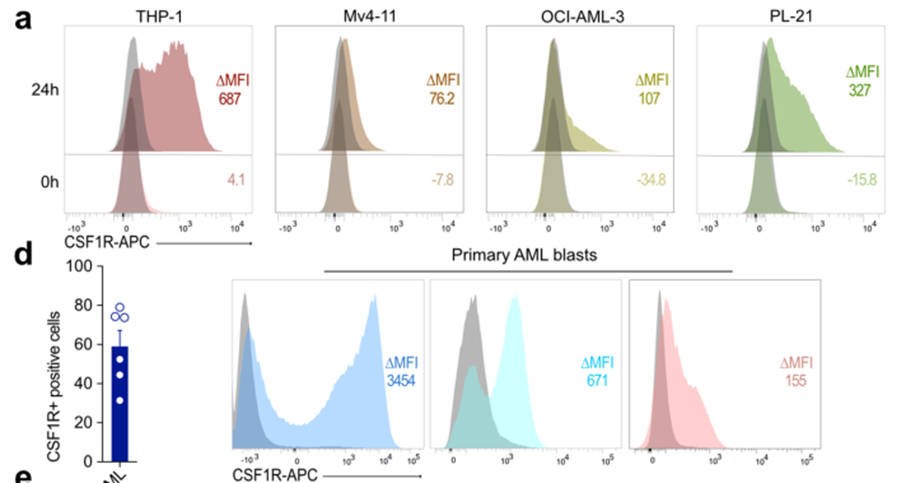
接下来,我们评估了原发性AML样本中的受体表达。到目前为止,CSF1R在AML原代细胞上的表达被认为局限于“AML支持细胞”或仅局限于成熟的白血病细胞46。事实上,在分析解冻后立即冻结的骨髓样本表面CSF1R表达时,我们无法通过流式细胞术检测到任何可测量的受体表达(图5a,b)。然而,当原代AML细胞在MS-5小鼠骨髓基质细胞上共培养时(扩展数据图5b),我们观察到CSF1R表达强烈且依赖于时间的增加(图5a,b)。
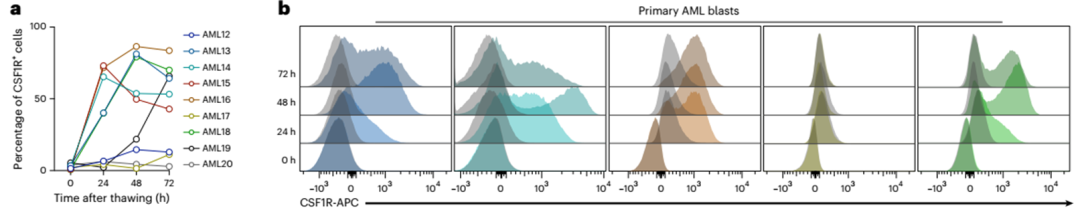
我们假设这些表面CSF1R表达的差异很可能是由于冻融过程中受体下调所致。为了探究这一点,我们分析了冻融循环后AML细胞系受体的表达。与AML原代细胞的结果相似,CSF1R在解冻后无法直接检测到,但在培养24 - 48小时后恢复了高表达(扩展数据图5a)。为了进一步排除任何细胞培养产物,我们分析了在细胞因子丰富的培养基中培养的AML原代细胞表面受体的表达47(扩展数据图5c)。同样,CSF1R在恶性AML原代细胞培养后高表达(扩展数据图5d)。我们还证实了CD86在AML原代细胞中的表达(图5c)。
我们的单细胞基因表达分析显示,CSF1R和CD86在恶性造血干细胞中的表达低于CD123和CD33内参基因。因此,我们分析了CSF1R和CD86在恶性hspc样细胞上的蛋白表达(扩展数据图5e-g)。CSF1R和CD86均在恶性hspc样细胞上表达,在这些细胞类型之间表达无差异(Extended Data Fig. 5f,g),说明这些细胞上靶抗原的保守表达。
接下来,我们将原代AML样本与CAR-T细胞共培养,并通过流式细胞仪检测其特异性裂解。hCSF1R和CD86 CART特异性地溶解了原发性AML样本,与低E:T比值的CD33 CART相比(图5d)。为了反映AML的遗传异质性,七个不同细胞遗传学的AML原始标本被用于体外检测。为了探索新的抗靶向CAR是否可以引入AML患者的T细胞中,我们将抗hcsf1r CAR构建物转导到AML患者的T细胞中(图5e)。然后将人源hCSF1R CART与自体AML原代细胞共培养,导致原代样本的强效裂解(图5f)。
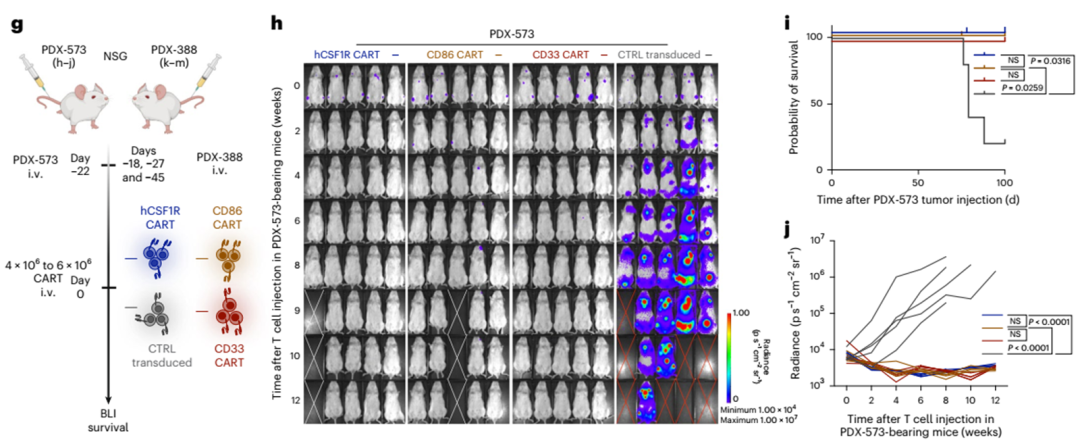
为了证明hCSF1R和CD86 CAR-T在更相关的体内模型中的有效性,我们将细胞遗传学上不同的人源异种移植(PDX)模型移植到小鼠体内,并用各自的CAR-T细胞治疗它们。首先,我们选择了PDX-573模型,该模型来源于一名患有高风险细胞遗传学的复发AML患者(European LeukemiaNet 2017,不良预后;详细特征见扩展数据表1)。三周后,我们注射hCSF1R、CD86、CD33或CD19 CART(图5g-j)。所有CAR-T细胞都是非常有效的,在所有治疗小鼠中抑制肿瘤生长(5 / 5;图5h-j)。
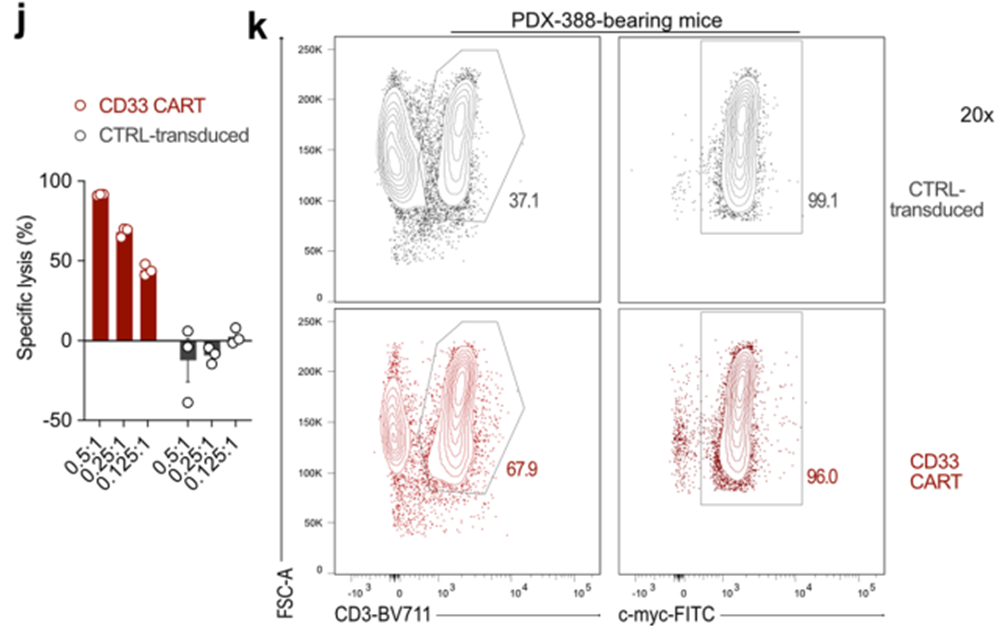
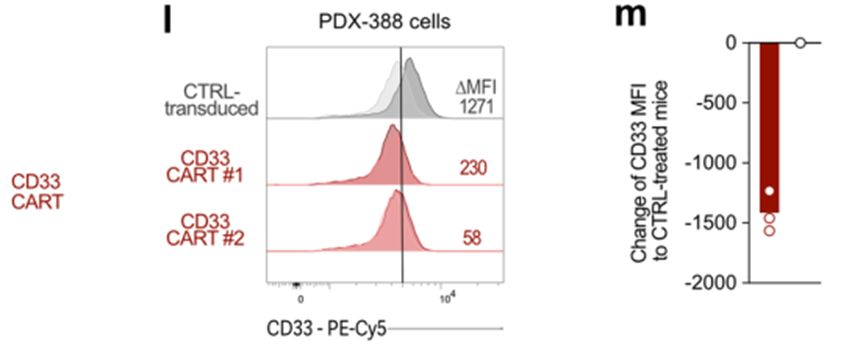
接下来,我们检测了hCSF1R、CD86和CD33 CART在PDX-388中的疗效,PDX-388来源于一名初诊时患有KMT2A重排的AML患者(European leukemia anet 2017,不良预后;图5k-m)。值得注意的是,CSF1R在PDX388样品上的表达符合上述模式;细胞解冻后,CSF1R在PDX-388细胞上不表达,但在体外培养至少24小时后可检测到(扩展数据图5h),在对照处理的PDX-388小鼠的体内骨髓切片上也可检测到(扩展数据图5i)。hCSF1R和CD86 CART在所有治疗的小鼠中诱导了85 d的持续缓解(10 / 10的CSF1R CART和3 / 3的CD86 CART中的CR;图5k-m)。
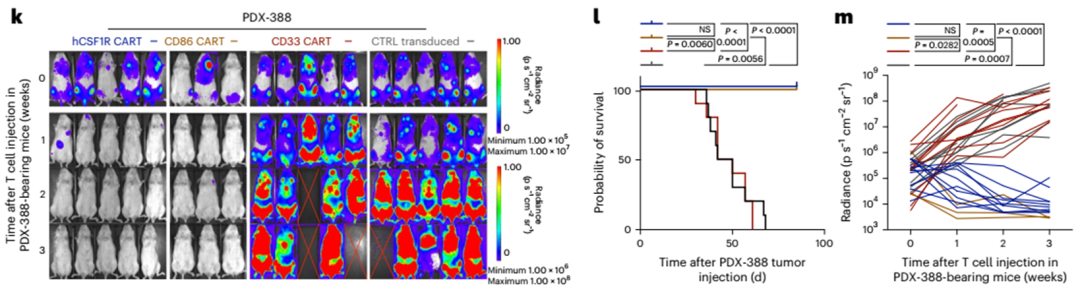
有趣的是,在这个模型中,CD33 CART完全不能控制所有小鼠的肿瘤生长(cr0 = 10)。我们排除了体外制造CD33 CART的失败(扩展数据图5j)。此外,在一个单独的队列中,我们验证了CD33 CAR在治疗小鼠的循环中存在(扩展数据图5k,左),并在细胞表面表达了CAR(扩展数据图5k,右)。体外流式细胞术检测PDX-388细胞上的CD33显示,与CD19 CART处理的小鼠相比,CD33 CART处理的小鼠PDX细胞上的CD33表面表达显著降低(扩展数据图5l,m)。因此,CD33 CART无法控制肿瘤负荷很可能是由于PDX-388母细胞表面CD33表达下调所致。然而,其具体的生物学机制尚不清楚,还需要进一步的研究。
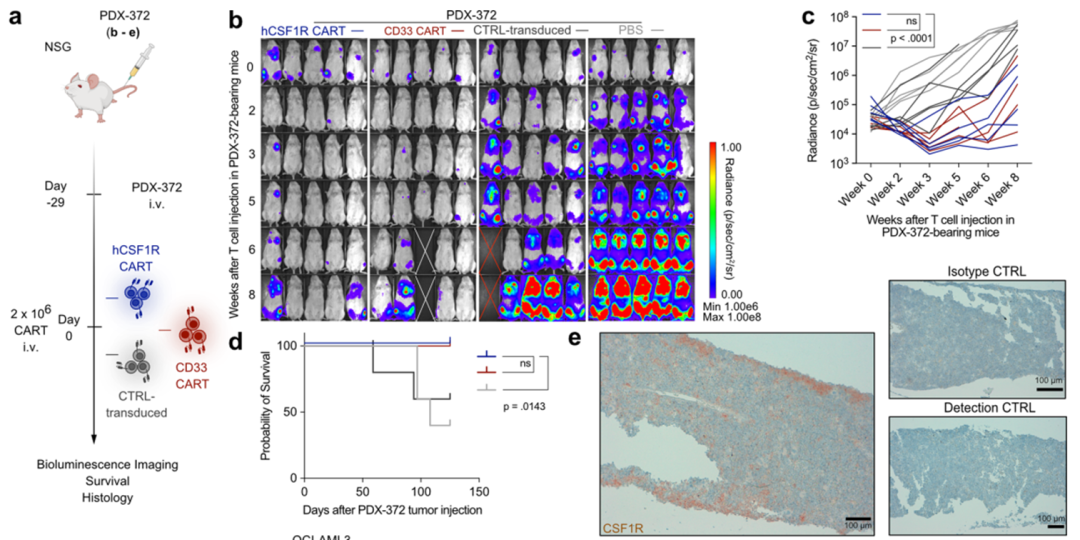
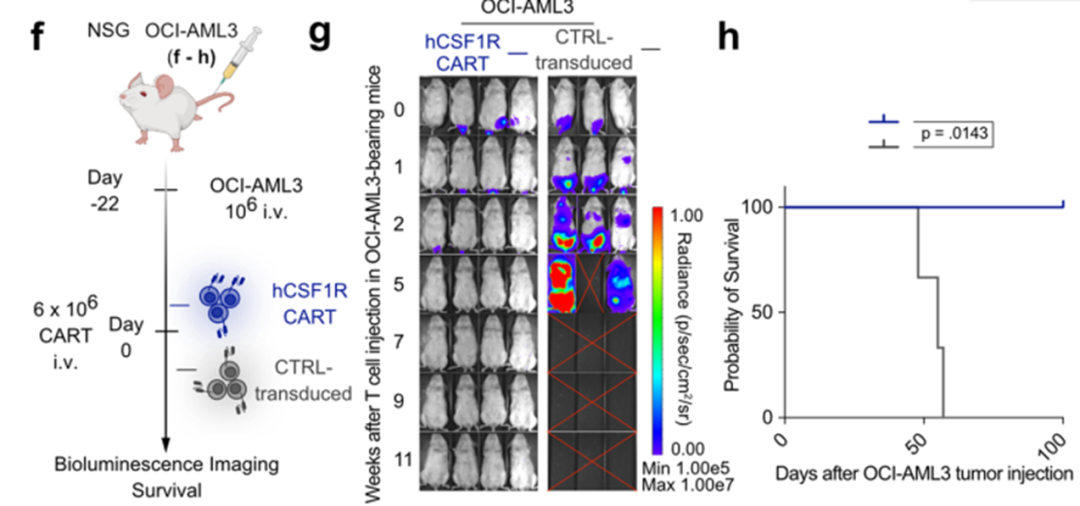
为了明确地在体内验证hCSF1R CART的潜力,我们使用了第三个PDX模型(PDX-372;扩展数据图6a-e)和第三种细胞系异种移植模型(OCI-AML3;扩展数据图6f-h)PDX-372样本再次来自于一个患有高危细胞遗传学和TP53突变的复发AML患者(扩展数据表1)。此外,为了创建更具挑战性的模型,我们将减少数量的CAR-T细胞转移到携带PDX-372的小鼠中(扩展数据图6a)。。hCSF1R CART抑制了5只小鼠中3只的AML生长。检测到的BLI信号在hCSF1R和CD33 CART之间没有变化(扩展数据图6b,c)。如前所述的PDX-388,免疫组化分析显示CSF1R在体内的PDX细胞上高表达(扩展数据图6e)。将hCSF1R CART转移到OCI-AML3荷瘤小鼠中同样有效(扩展数据图6f-h)。
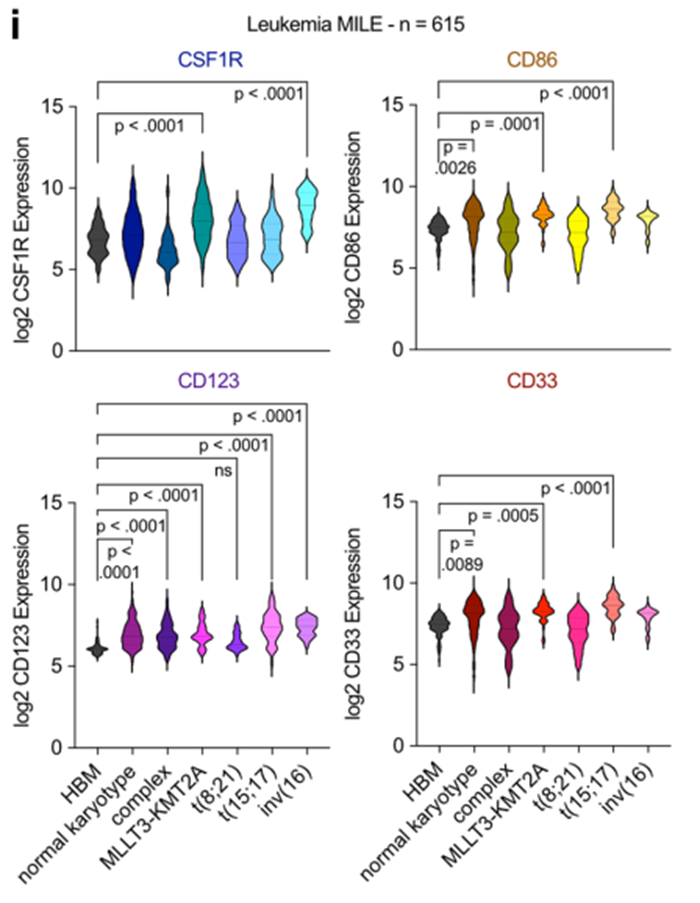
为了更好地理解CSF1R和CD86在AML复杂分子结构中的表达模式,以及在不同AML亚型中可能存在的不同表达模式,我们使用了一个公开的大规模数据集(白血病MILE研究),并将CSF1R和CD86的表达与CD123和CD33内参基因进行了比较。与CD33相似,CSF1R和CD86在不同亚型中广泛表达,KMT2A::MLLT3 (MLL::AF9)、t(15;17)和inv(16)突变的AML中表达最高(扩展数据图6i)。
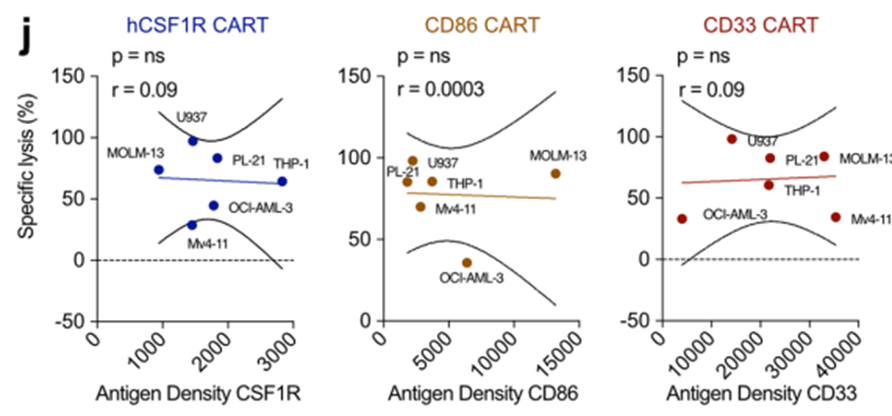
考虑到我们在整个研究中使用的各种体外和体内模型,我们下一步试图研究我们是否可以确定一个有效的CAR-T细胞治疗AML的抗原阈值。然而,我们没有观察到流式细胞术测量的抗原位点密度与CAR-T细胞对任何测试抗原的裂解能力之间的相关性(扩展数据图6j)。
总之,使用三种不同的、细胞遗传学上不同的PDX模型和三种细胞系异种移植模型,我们能够提供强有力的证据,证明新开发的抗靶向CAR-T细胞在体外和体内的功能。
结果6 hCSF1R和CD86 CART毒性分析
在验证其在恶性AML细胞上的表达后,我们接下来评估了CD34+ hspc上的靶抗原表达。使用流式细胞术,我们发现CSF1R和CD86在健康hspc中的表达低于CD33(图6a,b)。
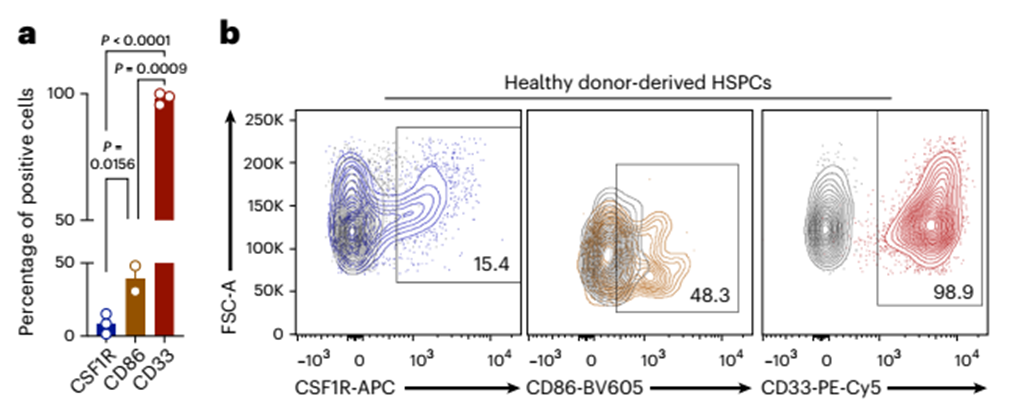
为了直接评估对hscs的毒性,我们将富含骨髓的CD34+细胞与hCSF1R、CD86和CD33 CART或未转导的T细胞共培养24小时(图6含有hCSF1R、CD86和CD33 CART或未转导的T细胞共培养24小时(图6c)。CD34+ hspc被CD33 CART特异地裂解(图6c,左)。同时,CD33 CART比hCSF1R或CD86 CART分泌更多的IFNγ进入共培养上清(图6c,右)。为了进一步验证这些结果,我们进行了常规的菌落形成单元(c.f.u)测定。与CD33 CART共培养时相比,hCSF1R CART共培养时的cfu .u -E和burst-forming unit (BFU)-E的菌落数更高,表明hCSF1R CART存在时干细胞的存活率更高(图6d)。重要的是,与hCSF1R CART或未转导的T细胞共培养的hspc的菌落计数没有变化(图6d)。
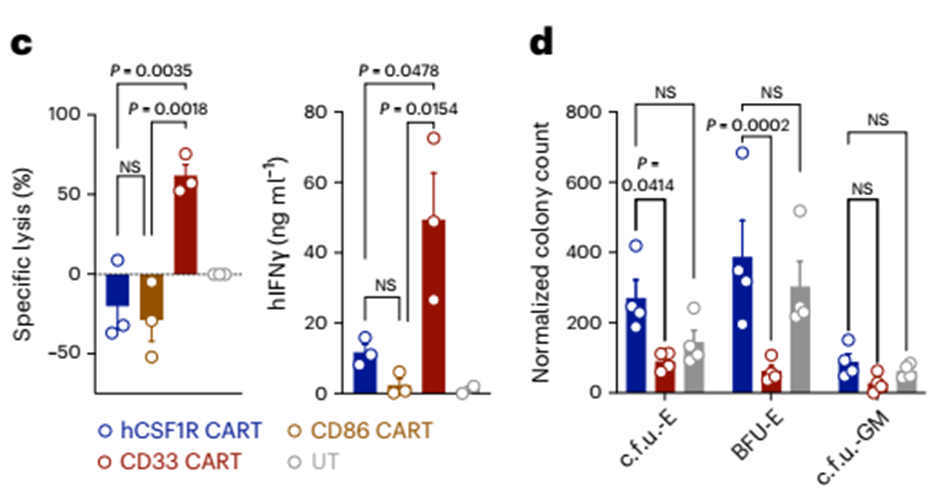
接下来,我们分析了目标抗原在健康人类骨髓供体样本(HD样本;图6e、f)。同样,表面CSF1R的表达至少在培养24 h后才能检测到(图6e),但其表达仍低于CD86或CD33(图6f)。将hCSF1R和CD33 CART或未转导的T细胞与HD样本共培养,发现HD样本的裂解率更高(图6g,左),CD33 CART的IFNγ分泌增加(图6g,右)。HD样本的裂解和IFNγ分泌在hCSF1R CART和未转导的T细胞之间没有差异(图6g)。
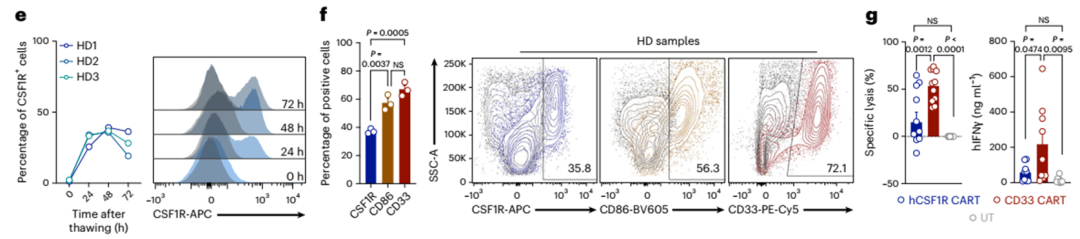
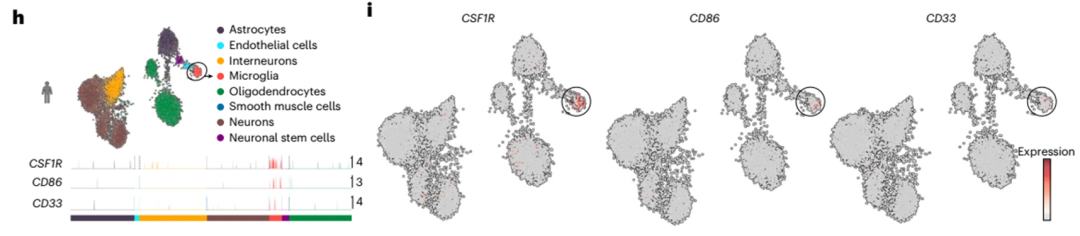
对单个人脑细胞的scRNA-seq分析证实了CSF1R在小胶质细胞中的表达(图6h,i)。在单细胞水平上,CSF1R在小胶质细胞中的表达高于CD86或CD33(图6h,i)。
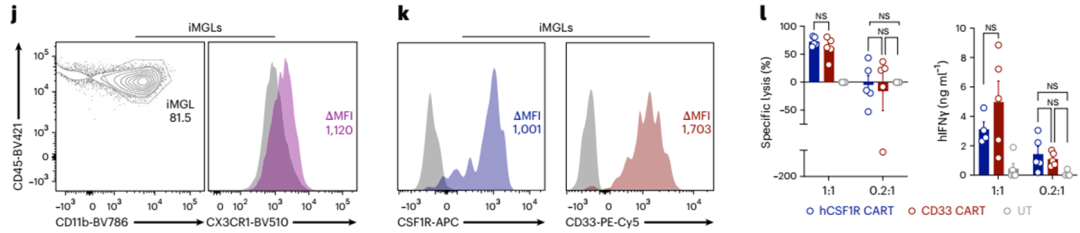
为了建立CAR-T细胞对人类小胶质细胞的毒性模型,我们生成诱导多能干细胞(iPSC)衍生的人类小胶质细胞样细胞(iMGLs)49,50,并验证其表型(图6j)。CSF1R和CD33都在iMGLs上高表达(图6k)。将人iMGLs与CSF1R CART、CD33 CART或未转导的T细胞共培养,结果表明,在E:T比值为1:1的情况下,两种CAR均可裂解人iMGLs(图6l,左)。在更多的生理E:T比值(0.2:1)下,CSF1R和CD33 CART都不能溶解人imgl,这与我们的体内数据一致(图6l,左)。IFNγ释放与流式细胞仪分析结果相似(图6l,右)。
上一篇急性心肌梗塞(AMI)外周血scRNA-Seq分析流程及结果梳理
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-12-17,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号