转录组分析—GSE200033二分组(去除异常值Vs未去除)
原创
转录组分析—GSE200033二分组(去除异常值Vs未去除)
原创
sheldor没耳朵
发布于 2024-07-31 18:26:56
发布于 2024-07-31 18:26:56
转录组分析—GSE200033二分组(去除异常值Vs未去除)
1 数据的读取与导入
1.1 文件下载
直接从GEO官网下载表达矩阵,临床信息表格(存放在Series Matrix File中),放在工作目录下;
表达矩阵有两个GSE200033_CountTable_BMDM.csv.gz,GSE200033_normCounts_BMDM.csv.gz(标准化后),区别如下,这里我们选择前者。
#查看二者的区别
dat <- fread("GSE200033_CountTable_BMDM.csv.gz")
dat1 <- fread("GSE200033_normCounts_BMDM.csv.gz")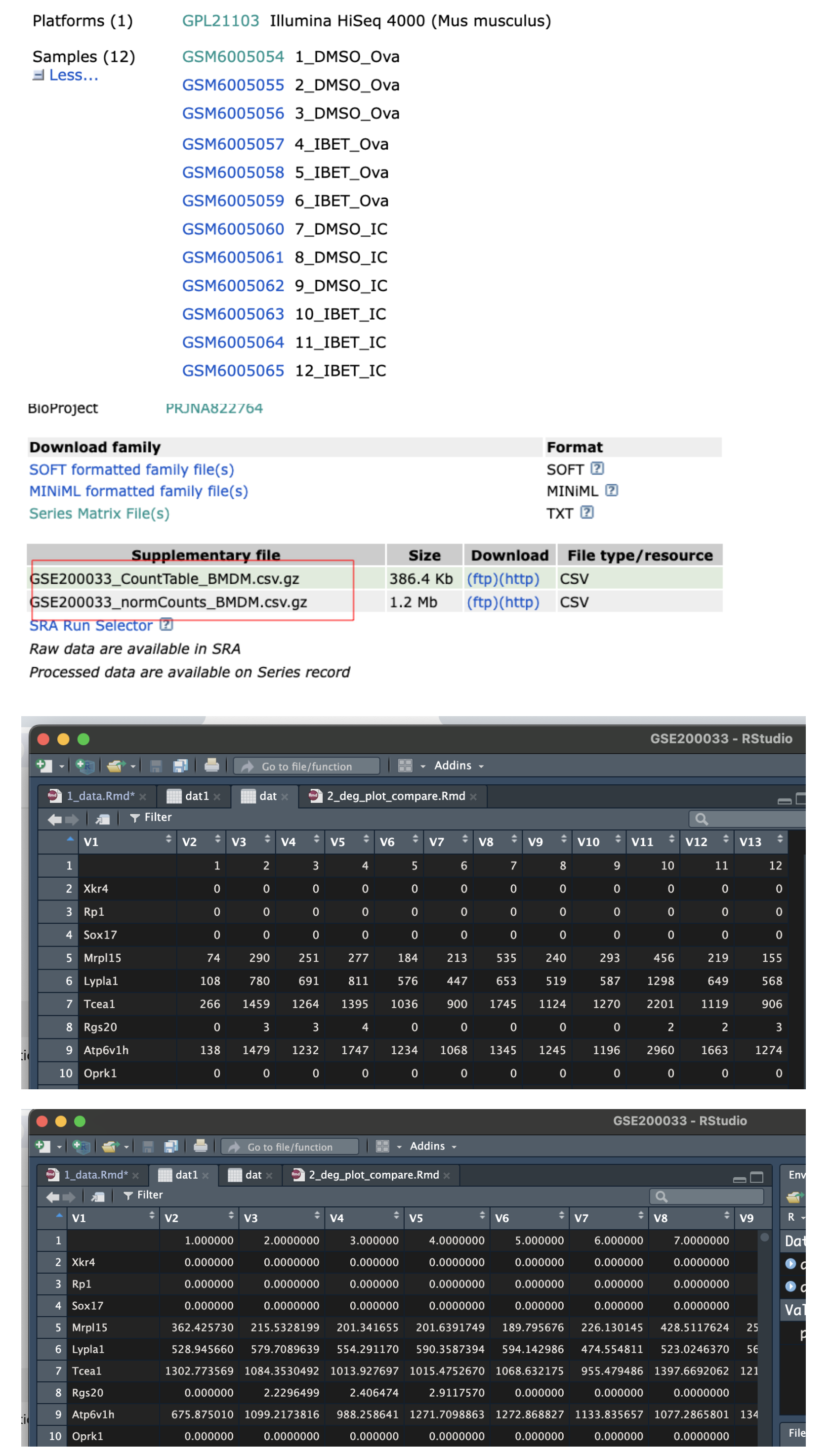
1.2 读取与整理
#项目名称
rm(list=ls())
proj = "GSE200033"
#表达矩阵
library(tinyarray)
library(data.table)
dat <- read.csv("GSE200033_CountTable_BMDM.csv.gz",header = T)
dim(dat)
#> [1] 24421 13
rownames(dat) <- dat$X
dat <- dat[,3:7]
colnames(dat) <- c("2_DMSO_Ova","3_DMSO_Ova",
"4_IBET_Ova","5_IBET_Ova","6_IBET_Ova")
#检查行名、列名
#rownames(dat)
colnames(dat)
#> [1] "2_DMSO_Ova" "3_DMSO_Ova" "4_IBET_Ova" "5_IBET_Ova" "6_IBET_Ova"
# 转换为整数矩阵
exp <-as.matrix(round(dat))
#临床信息在表头上不需要处理clinical占位置,无实际含义
clinical =1
nrow(exp)
#> [1] 24421
exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]
nrow(exp)
#> [1] 13705这样就把表达矩阵的形式转化为行名为基因名,列名为分组信息的名字。本意是做前6个GSM的比较,但是后续的PCA结果显示,样本1为离群值,因此去除了样本1后重新分析了一遍。
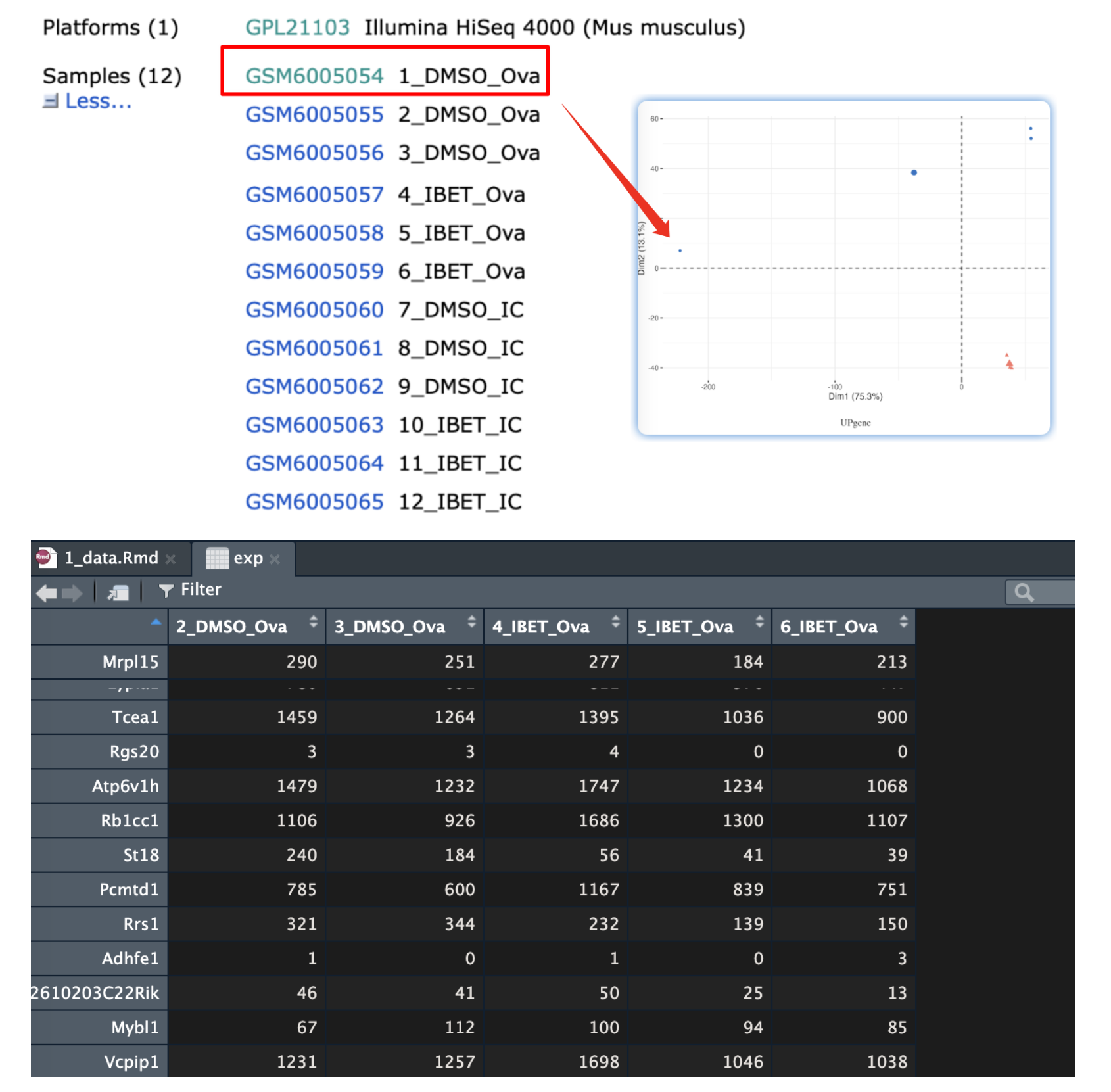
#分组信息的获取
library(stringr)
k = str_detect(colnames(exp),"DMSO");table(k)
Group = ifelse(k,"DMSO","IBET")
Group = factor(Group,levels = c("DMSO","IBET"));Group
data.frame(colnames(exp),Group)
#保存数据
save(exp,Group,proj,clinical,file = paste0(proj,".Rdata"))2 差异基因
三大R包的差异基因分析
rm(list = ls())
load("GSE200033.Rdata")
table(Group)
#> Group
#> DMSO IBET
#> 2 3
#deseq2----
library(DESeq2)
colData <- data.frame(row.names =colnames(exp),
condition=Group)
#注意事项:如果多次运行,表达矩阵改动过的话,需要从工作目录下删除下面if括号里的文件
if(!file.exists(paste0(proj,"_dd.Rdata"))){
dds <- DESeqDataSetFromMatrix(
countData = exp,
colData = colData,
design = ~ condition)
dds <- DESeq(dds)
save(dds,file = paste0(proj,"_dd.Rdata"))
}
load(file = paste0(proj,"_dd.Rdata"))
class(dds)
#> [1] "DESeqDataSet"
#> attr(,"package")
#> [1] "DESeq2"
res <- results(dds, contrast = c("condition",rev(levels(Group))))
#constrast
c("condition",rev(levels(Group)))
#> [1] "condition" "IBET" "DMSO"
class(res)
#> [1] "DESeqResults"
#> attr(,"package")
#> [1] "DESeq2"
DEG1 <- as.data.frame(res)
library(dplyr)
DEG1 <- arrange(DEG1,pvalue)
DEG1 = na.omit(DEG1)
head(DEG1)
#> baseMean log2FoldChange lfcSE stat pvalue padj
#> Kctd12 7068.6436 3.154592 0.1539593 20.48978 2.655694e-93 3.231183e-89
#> Ccr5 1068.4898 -4.493158 0.2597624 -17.29718 4.939449e-67 3.004914e-63
#> H3f3b 6887.4175 2.089414 0.1220509 17.11920 1.067240e-65 4.328371e-62
#> Gpr183 287.3395 -3.565865 0.2232215 -15.97456 1.922093e-57 5.846526e-54
#> Brd2 8184.5773 2.496878 0.1594698 15.65737 2.958994e-55 7.200417e-52
#> Plau 3264.8888 -2.561643 0.1640528 -15.61475 5.777151e-55 1.171510e-51
#添加change列标记基因上调下调
logFC_t = 1
pvalue_t = 0.05
k1 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange < -logFC_t);table(k1)
#> k1
#> FALSE TRUE
#> 11295 872
k2 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange > logFC_t);table(k2)
#> k2
#> FALSE TRUE
#> 11636 531
DEG1$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG1$change)
#>
#> DOWN NOT UP
#> 872 10764 531
head(DEG1)
#> baseMean log2FoldChange lfcSE stat pvalue padj
#> Kctd12 7068.6436 3.154592 0.1539593 20.48978 2.655694e-93 3.231183e-89
#> Ccr5 1068.4898 -4.493158 0.2597624 -17.29718 4.939449e-67 3.004914e-63
#> H3f3b 6887.4175 2.089414 0.1220509 17.11920 1.067240e-65 4.328371e-62
#> Gpr183 287.3395 -3.565865 0.2232215 -15.97456 1.922093e-57 5.846526e-54
#> Brd2 8184.5773 2.496878 0.1594698 15.65737 2.958994e-55 7.200417e-52
#> Plau 3264.8888 -2.561643 0.1640528 -15.61475 5.777151e-55 1.171510e-51
#> change
#> Kctd12 UP
#> Ccr5 DOWN
#> H3f3b UP
#> Gpr183 DOWN
#> Brd2 UP
#> Plau DOWN
#edgeR----
library(edgeR)
dge <- DGEList(counts=exp,group=Group)
dge$samples$lib.size <- colSums(dge$counts)
dge <- calcNormFactors(dge)
design <- model.matrix(~Group)
dge <- estimateGLMCommonDisp(dge, design)
dge <- estimateGLMTrendedDisp(dge, design)
dge <- estimateGLMTagwiseDisp(dge, design)
fit <- glmFit(dge, design)
fit <- glmLRT(fit)
DEG2=topTags(fit, n=Inf)
class(DEG2)
#> [1] "TopTags"
#> attr(,"package")
#> [1] "edgeR"
DEG2=as.data.frame(DEG2)
head(DEG2)
#> logFC logCPM LR PValue FDR
#> Sema4d -2.819340 7.941342 422.0946 8.541659e-94 1.170634e-89
#> Ccr5 -4.328710 6.792529 409.8625 3.926969e-91 2.690956e-87
#> Plau -2.688427 8.719388 353.1610 8.685305e-79 3.967737e-75
#> Gpr183 -3.526972 5.049178 267.3149 4.366338e-60 1.496017e-56
#> Sat1 2.638522 8.577100 266.8574 5.493330e-60 1.505722e-56
#> Mt1 2.544215 7.199551 263.5216 2.930275e-59 6.693237e-56
k1 = (DEG2$PValue < pvalue_t)&(DEG2$logFC < -logFC_t)
k2 = (DEG2$PValue < pvalue_t)&(DEG2$logFC > logFC_t)
DEG2$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
head(DEG2)
#> logFC logCPM LR PValue FDR change
#> Sema4d -2.819340 7.941342 422.0946 8.541659e-94 1.170634e-89 DOWN
#> Ccr5 -4.328710 6.792529 409.8625 3.926969e-91 2.690956e-87 DOWN
#> Plau -2.688427 8.719388 353.1610 8.685305e-79 3.967737e-75 DOWN
#> Gpr183 -3.526972 5.049178 267.3149 4.366338e-60 1.496017e-56 DOWN
#> Sat1 2.638522 8.577100 266.8574 5.493330e-60 1.505722e-56 UP
#> Mt1 2.544215 7.199551 263.5216 2.930275e-59 6.693237e-56 UP
table(DEG2$change)
#>
#> DOWN NOT UP
#> 1065 11922 718
#limma----
library(limma)
dge <- edgeR::DGEList(counts=exp)
dge <- edgeR::calcNormFactors(dge)
design <- model.matrix(~Group)
v <- voom(dge,design, normalize="quantile")
fit <- lmFit(v, design)
fit= eBayes(fit)
DEG3 = topTable(fit, coef=2, n=Inf)
DEG3 = na.omit(DEG3)
k1 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC < -logFC_t)
k2 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC > logFC_t)
DEG3$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))
table(DEG3$change)
#>
#> DOWN NOT UP
#> 866 12091 748
head(DEG3)
#> logFC AveExpr t P.Value adj.P.Val B change
#> Plau -2.768474 8.145581 -32.11575 3.588485e-09 4.485476e-05 11.72741 DOWN
#> Sema4d -2.858756 7.294563 -28.82385 7.899272e-09 4.485476e-05 11.05425 DOWN
#> Sat1 2.590141 8.099939 27.15005 1.221555e-08 4.485476e-05 10.70106 UP
#> Kctd12 3.115596 9.486043 26.75825 1.357953e-08 4.485476e-05 10.60613 UP
#> H3f3b 2.041197 9.744115 26.08128 1.636438e-08 4.485476e-05 10.43943 UP
#> Brd2 2.525475 9.850541 25.22432 2.086714e-08 4.766403e-05 10.21866 UP
tj = data.frame(deseq2 = as.integer(table(DEG1$change)),
edgeR = as.integer(table(DEG2$change)),
limma_voom = as.integer(table(DEG3$change)),
row.names = c("down","not","up")
);tj
#> deseq2 edgeR limma_voom
#> down 872 1065 866
#> not 10764 11922 12091
#> up 531 718 748
save(DEG1,DEG2,DEG3,Group,tj,file = paste0(proj,"_DEG.Rdata"))可视化
library(ggplot2)
library(tinyarray)
exp[1:4,1:4]
#> 2_DMSO_Ova 3_DMSO_Ova 4_IBET_Ova 5_IBET_Ova
#> Mrpl15 290 251 277 184
#> Lypla1 780 691 811 576
#> Tcea1 1459 1264 1395 1036
#> Rgs20 3 3 4 0
# cpm 去除文库大小的影响
dat = log2(cpm(exp)+1)
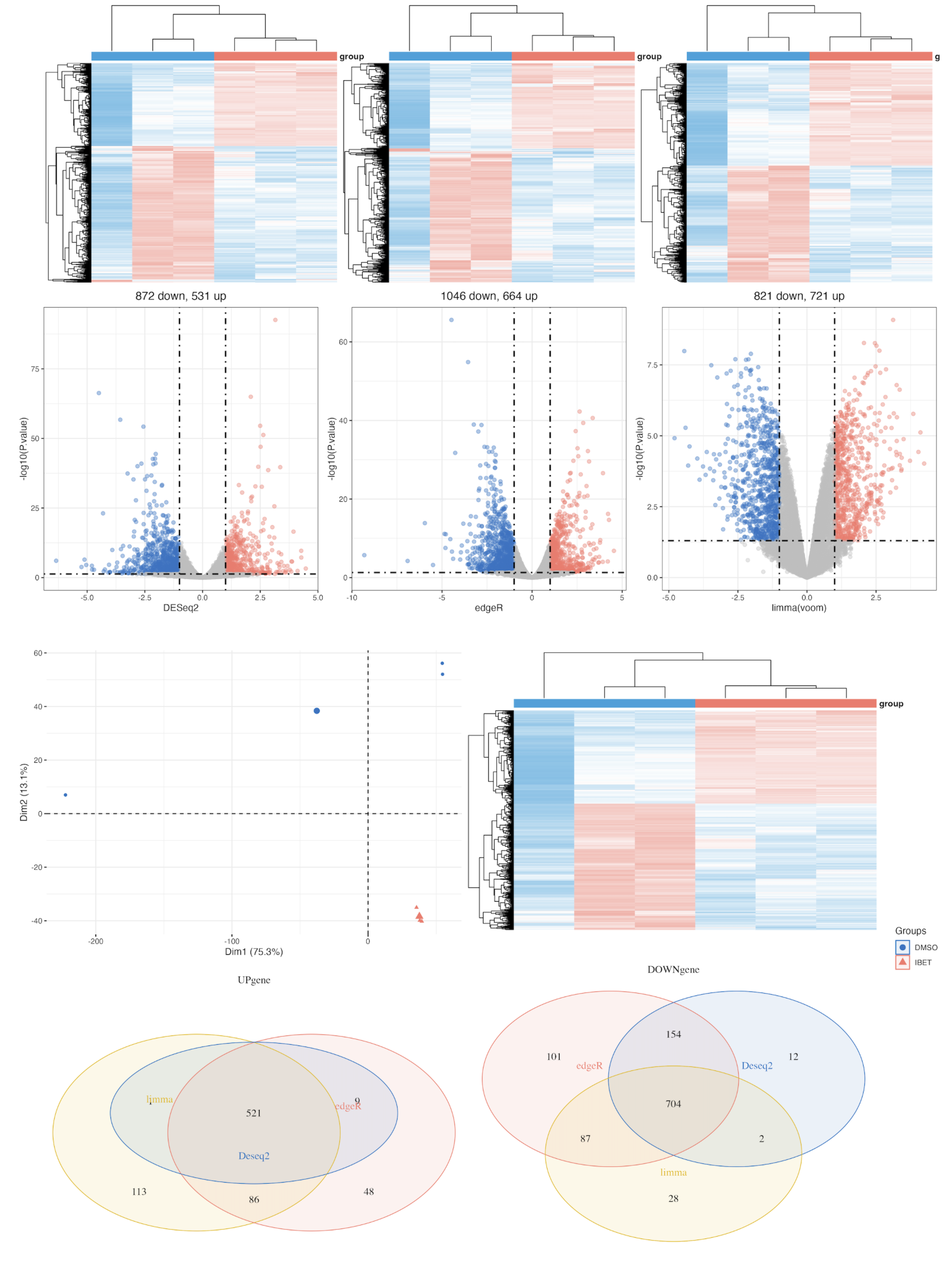
pca.plot = draw_pca(dat,Group);pca.plot
save(pca.plot,file = paste0(proj,"_pcaplot.Rdata"))
cg1 = rownames(DEG1)[DEG1$change !="NOT"]
cg2 = rownames(DEG2)[DEG2$change !="NOT"]
cg3 = rownames(DEG3)[DEG3$change !="NOT"]
h1 = draw_heatmap(dat[cg1,],Group)
h2 = draw_heatmap(dat[cg2,],Group)
h3 = draw_heatmap(dat[cg3,],Group)
v1 = draw_volcano(DEG1,pkg = 1,logFC_cutoff = logFC_t)
v2 = draw_volcano(DEG2,pkg = 2,logFC_cutoff = logFC_t)
v3 = draw_volcano(DEG3,pkg = 3,logFC_cutoff = logFC_t)
library(patchwork)
(h1 + h2 + h3) / (v1 + v2 + v3) +plot_layout(guides = 'collect') &theme(legend.position = "none")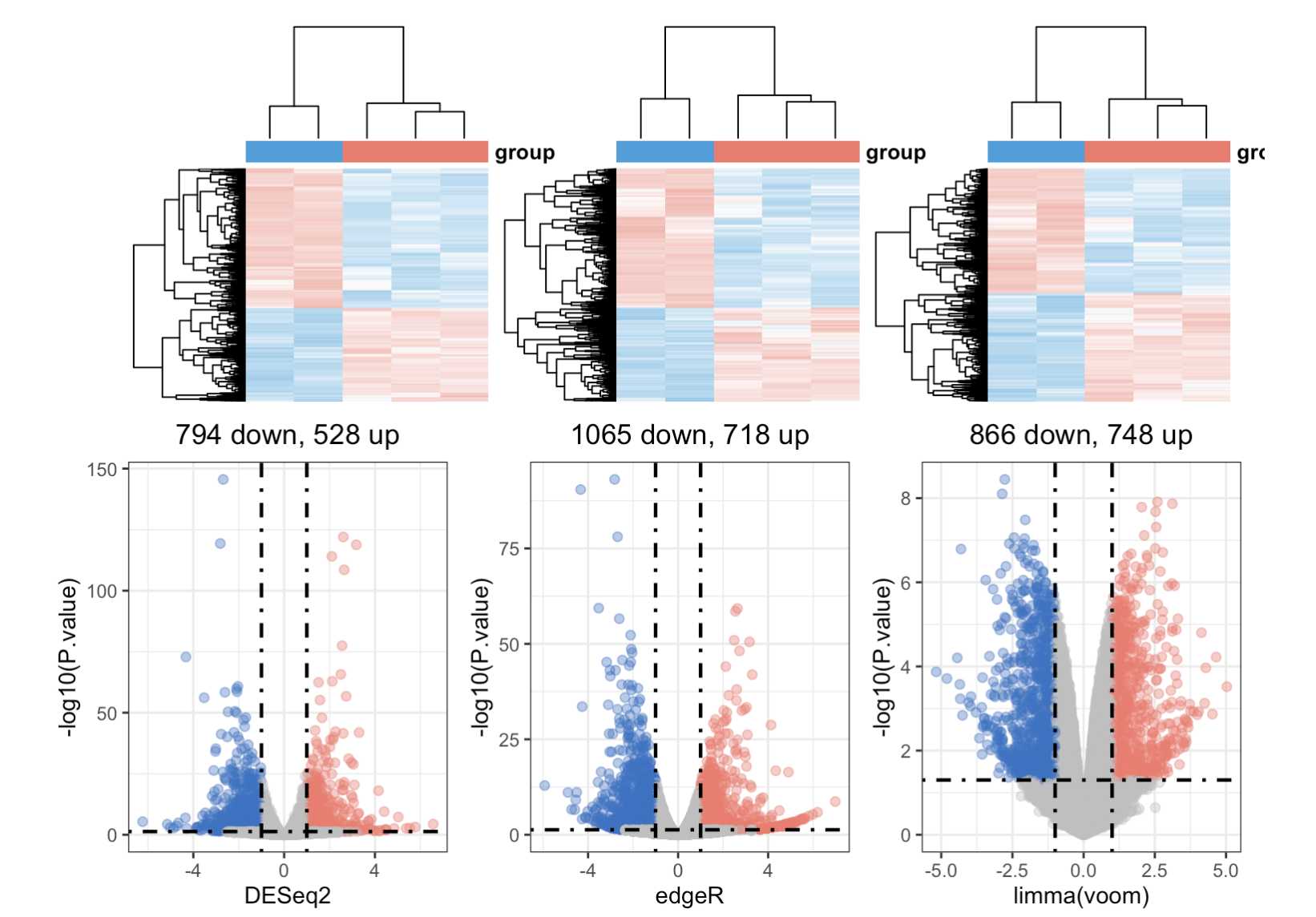
#三大R包差异基因对比
UP=function(df){
rownames(df)[df$change=="UP"]
}
DOWN=function(df){
rownames(df)[df$change=="DOWN"]
}
up = intersect(intersect(UP(DEG1),UP(DEG2)),UP(DEG3))
down = intersect(intersect(DOWN(DEG1),DOWN(DEG2)),DOWN(DEG3))
dat = log2(cpm(exp)+1)
hp = draw_heatmap(dat[c(up,down),],Group)
#上调、下调基因分别画维恩图
up_genes = list(Deseq2 = UP(DEG1),
edgeR = UP(DEG2),
limma = UP(DEG3))
down_genes = list(Deseq2 = DOWN(DEG1),
edgeR = DOWN(DEG2),
limma = DOWN(DEG3))
up.plot <- draw_venn(up_genes,"UPgene")
down.plot <- draw_venn(down_genes,"DOWNgene")
#维恩图拼图,终于搞定
library(patchwork)
#up.plot + down.plot
# 拼图
pca.plot + hp+up.plot +down.plot+ plot_layout(guides = "collect")
ggsave(paste0(proj,"_heat_ve_pca.png"),width = 15,height = 10)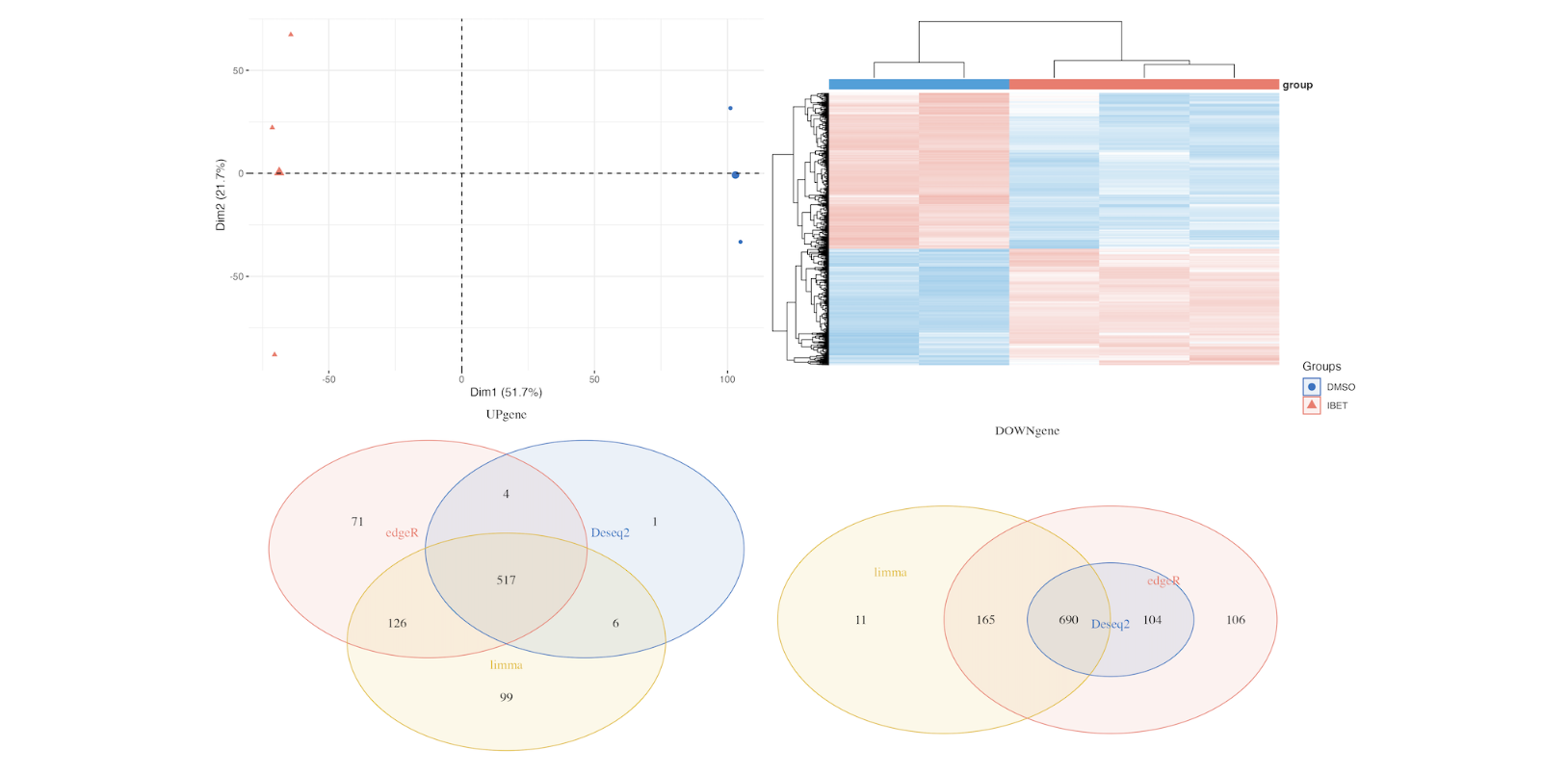
未去除异常样本的图如下
3 基因富集
取三个R包的交集差异基因做富集分析,注意应该用鼠源的数据库org.Mm.eg.db,kegg中organism = 'mmu',Go中OrgDb= org.Mm.eg.db。
rm(list = ls())
library(clusterProfiler)
library(ggthemes)
library(org.Mm.eg.db)
library(dplyr)
library(ggplot2)
library(stringr)
library(enrichplot)
#(1)输入数据
load("GSE200033_DEG.Rdata")
library(tinyarray)
g = intersect_all(rownames(DEG1)[DEG1$change!="NOT"],
rownames(DEG2)[DEG2$change!="NOT"],
rownames(DEG3)[DEG3$change!="NOT"])
output <- bitr(g,
fromType = 'SYMBOL',
toType = 'ENTREZID',
OrgDb = 'org.Mm.eg.db')
gene_diff = output$ENTREZID
#(2)富集
ekk <- enrichKEGG(gene = gene_diff,
organism = 'mmu')
ekk <- setReadable(ekk,
OrgDb = org.Mm.eg.db,
keyType = "ENTREZID")
ego <- enrichGO(gene = gene_diff,
OrgDb= org.Mm.eg.db,
ont = "ALL",
readable = TRUE)
#setReadable和readable = TRUE都是把富集结果表格里的基因名称转为symbol
class(ekk)
#> [1] "enrichResult"
#> attr(,"package")
#> [1] "DOSE"
#(3)可视化
p1 <- barplot(ekk)
p2 <- dotplot(ekk)
p1 + p2
ggsave(paste0("GSE200033_kegg.png"),width = 15,height = 10)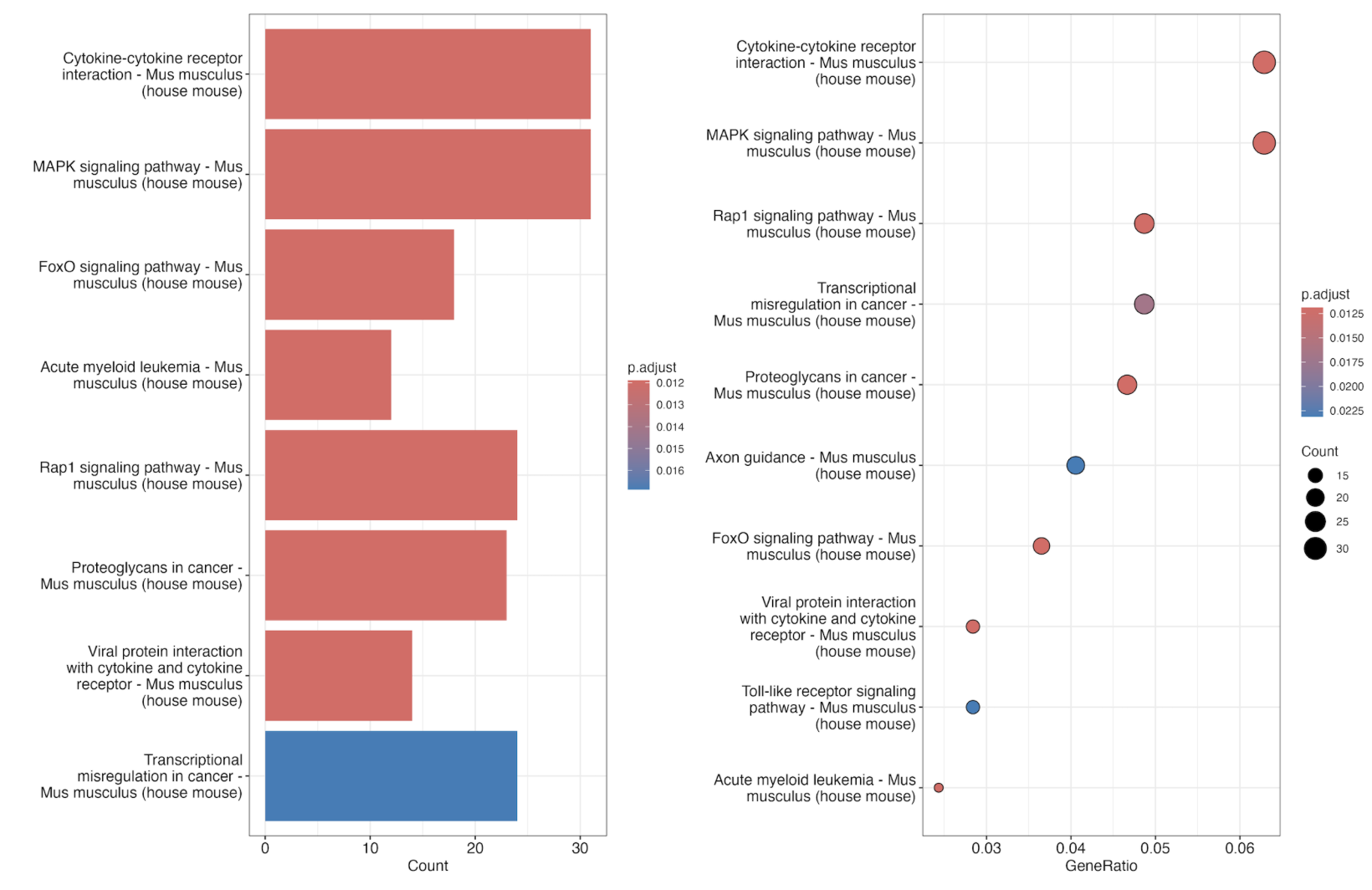
p3 <- barplot(ego, split = "ONTOLOGY") +
facet_grid(ONTOLOGY ~ ., space = "free_y",scales = "free_y")
p4 <- dotplot(ego, split = "ONTOLOGY") +
facet_grid(ONTOLOGY ~ ., space = "free_y",scales = "free_y")
p3+p4
ggsave(paste0("GSE200033_go.png"),width = 15,height = 13)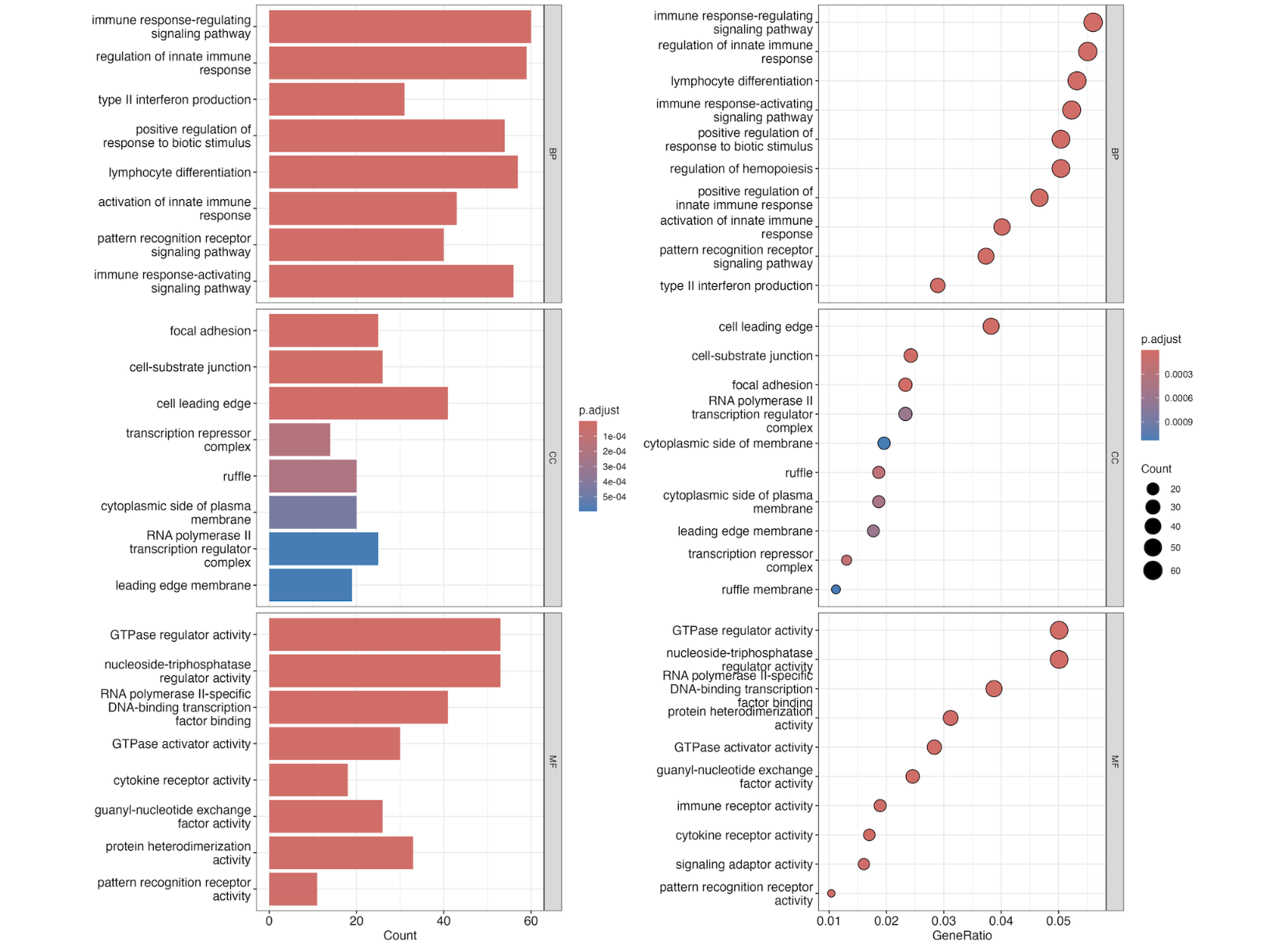
未去除异常样本的富集分析图如下
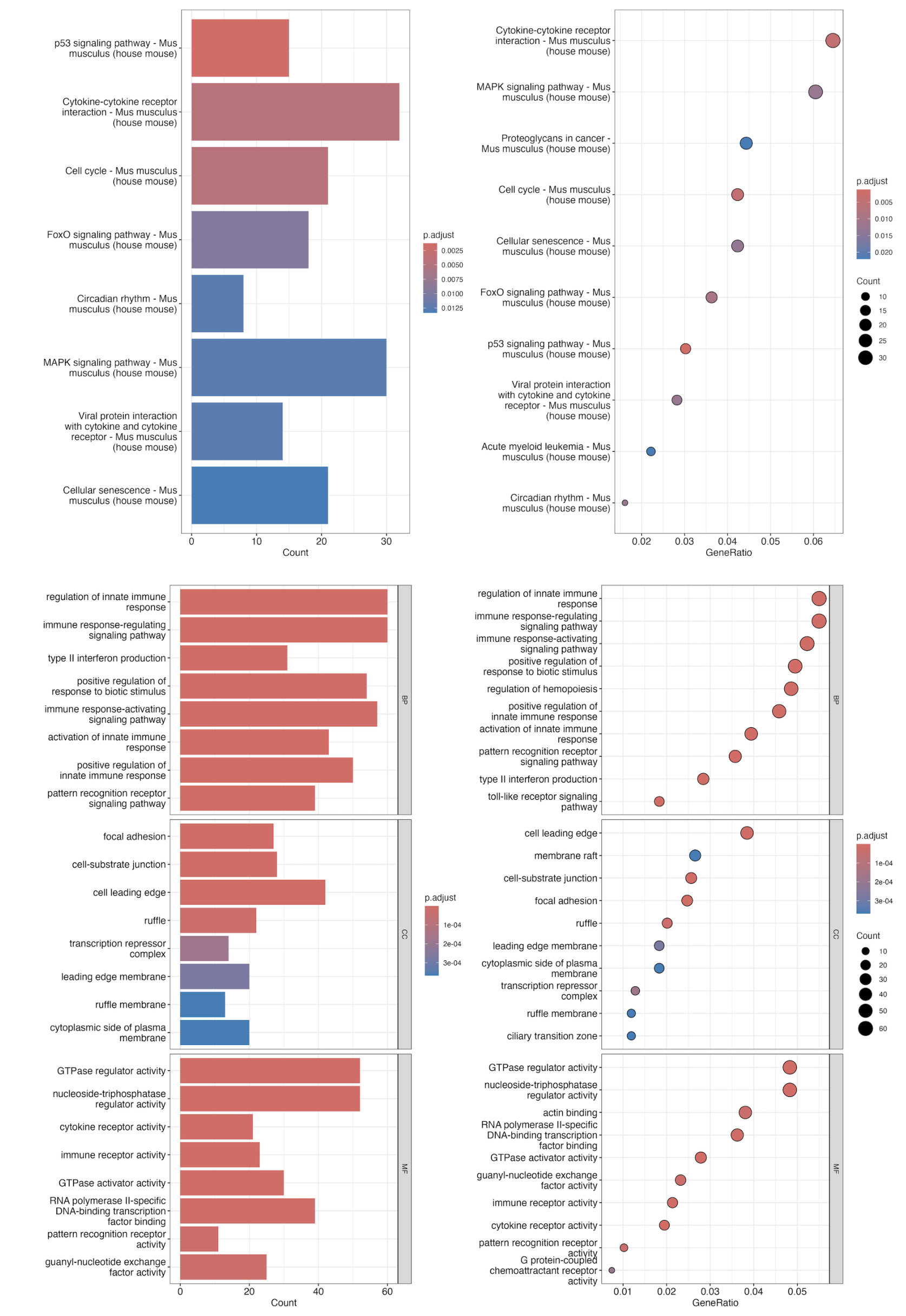
4 去除异常值 Vs 未去除异常值
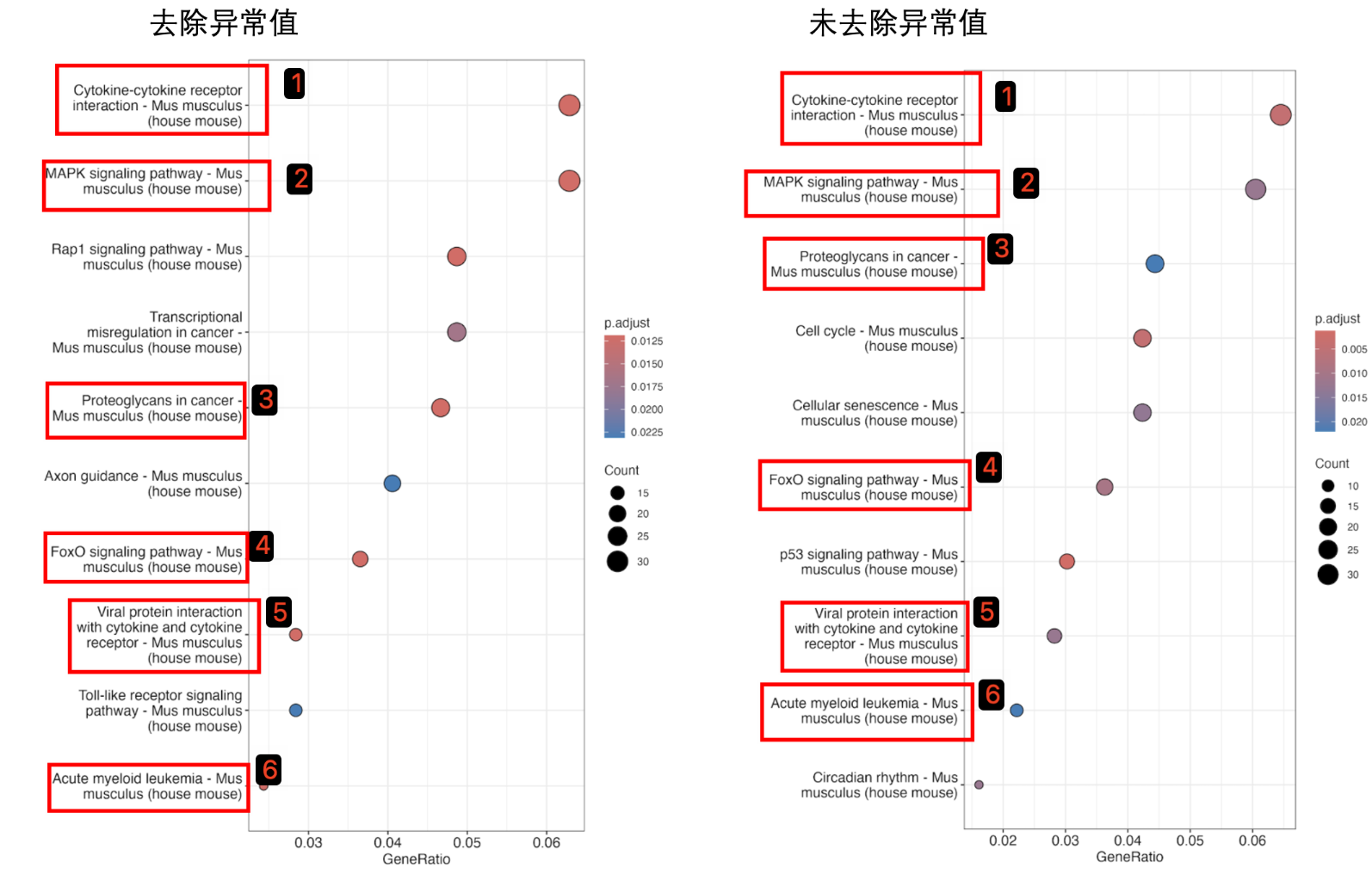
KEGG
Go
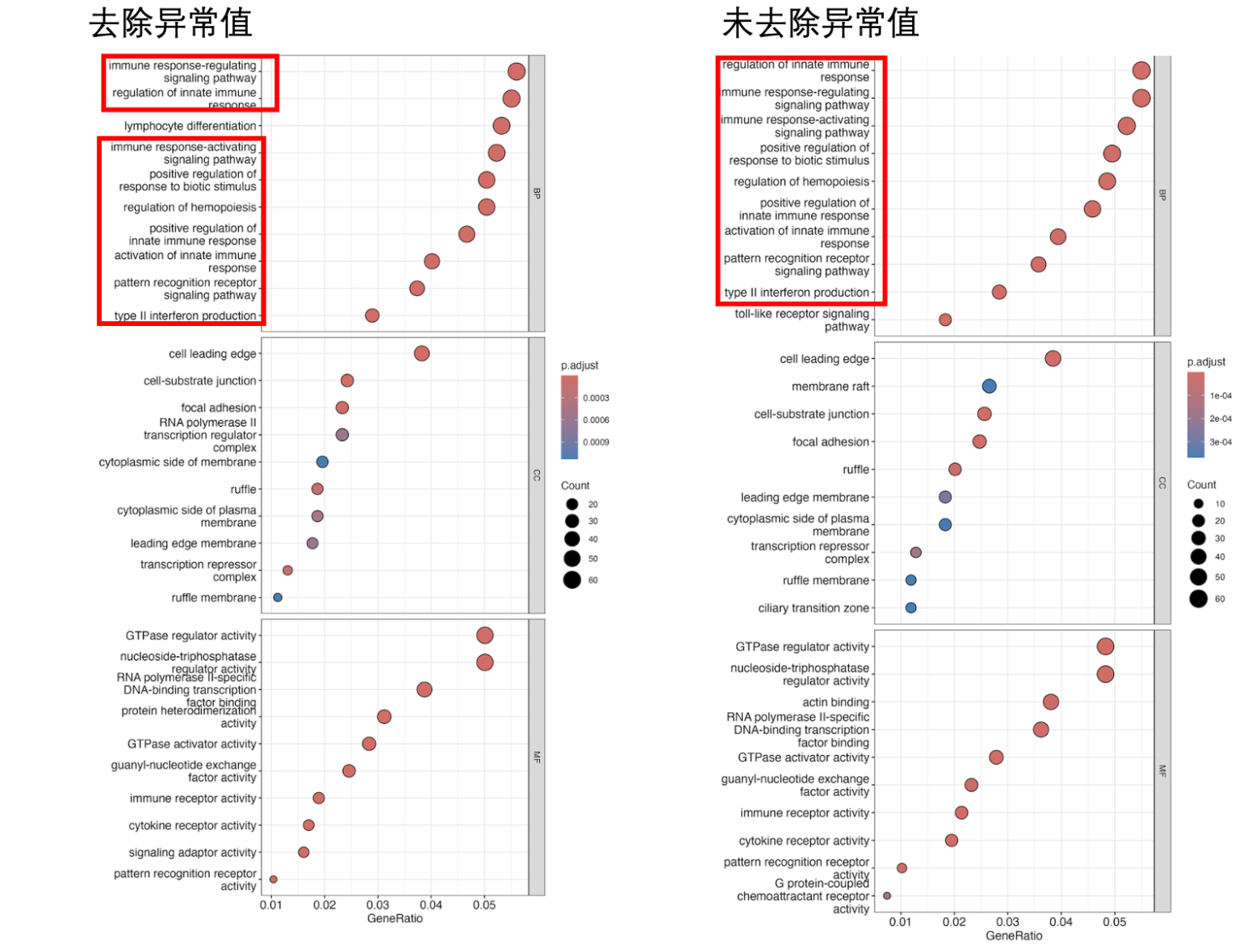
由此可以看出异常值去除与否,二者差异基因富集的通路并非相同。
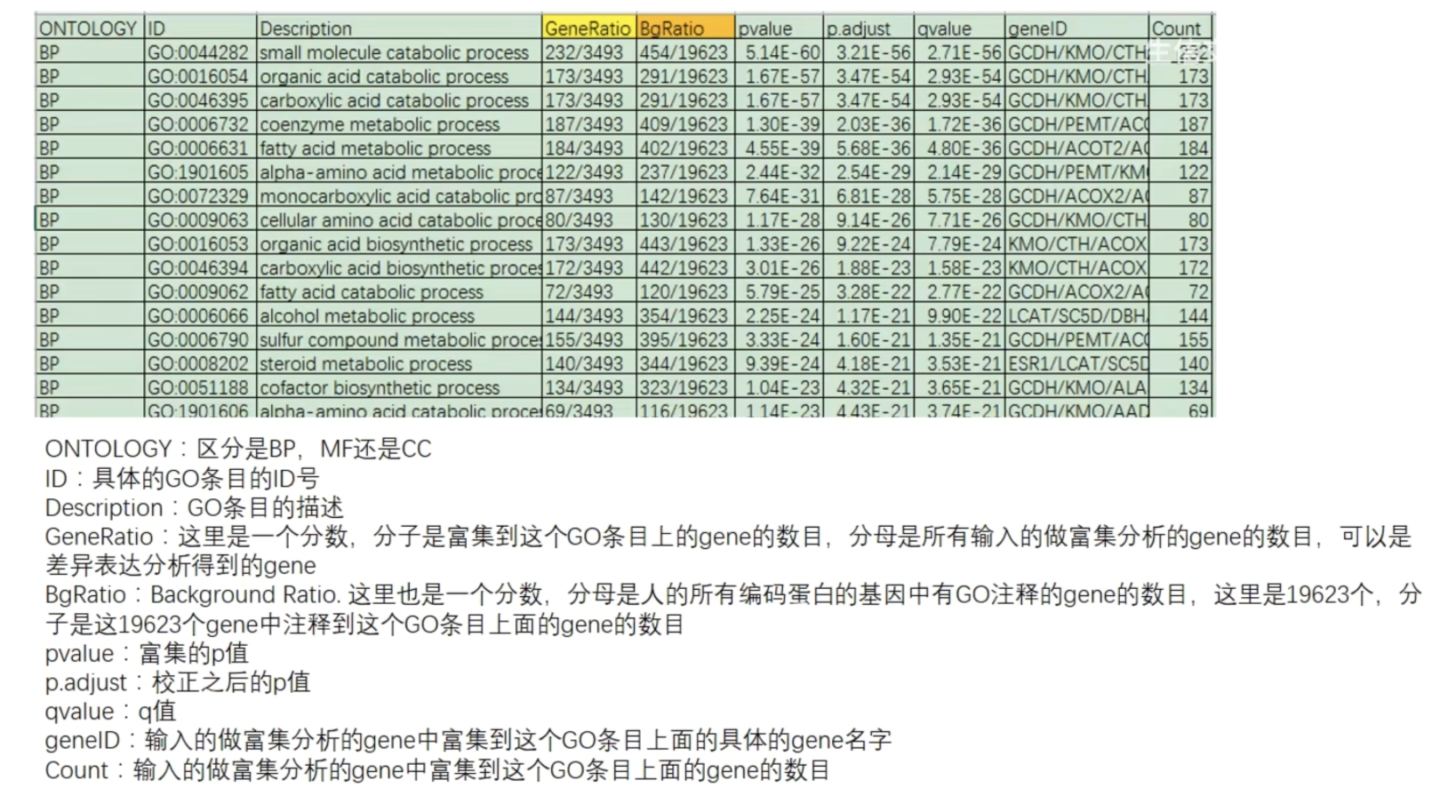
PS:补充知识
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号