月更单细胞图表复现-文献1-第四和五集


我们组建了一个月更的单细胞图表复现交流群,目前是进行到了第二个文献:文献复现-单细胞揭示新辅助治疗后NSCLC的免疫微环境变化,视频也剪辑好了在b站:

但是我们的文字版推文还在第一篇文献,前面已经分享了3个:胃癌单细胞数据集GSE163558复现(二):Seurat V5标准流程,接下来是图表美化和单细胞亚群比例探讨:
第四集:图表美化
背景介绍
绘图质量一定程度上会直接影响文章的发表。站在审稿人的角度,好看的图会令人赏心悦目,不好看的图会成为巨大的减分项。好看的图往往逻辑清晰、布局合理、配色协调。
逻辑清晰与否主要体现在机制/流程图上。这张图是全文的重中之重,是作者科研能力、绘图水平和文章质量的集中体现。布局是否合理大图和小图都会涉及。小图需要我们去不断的调整坐标轴、标签、文字的位置、大小和粗细,还有图片的透明度、分组的顺序等等等等。配色协调考验的是绘图人的审美水平。笔者是直男审美,在学习R绘图的时候,数据和代码的问题往往能解决,但就是画的一手丑图。

特别是配色一言难尽,只能找爱逛街的师妹帮忙配色,或者去找高分文章的配色然后直接copy。由于是审美不在线,因此本系列所有的颜色代码,我就老老实实的copy。
看看第三期的图,配色是不是看着很舒服:

2.可视化
在分群注释之后,我们可以使用DimPlot、FeaturePlot、DoHeatmap、DotPlot等多种函数对细胞或基因可视化。
在绘图之前,我们首先创建新的工作目录,并读取第三期亚群注释后的数据:
dir.create("4-plot")
setwd('4-plot/')
sce.all=readRDS( "../3-Celltype/sce_celltype.rds")
sce.all
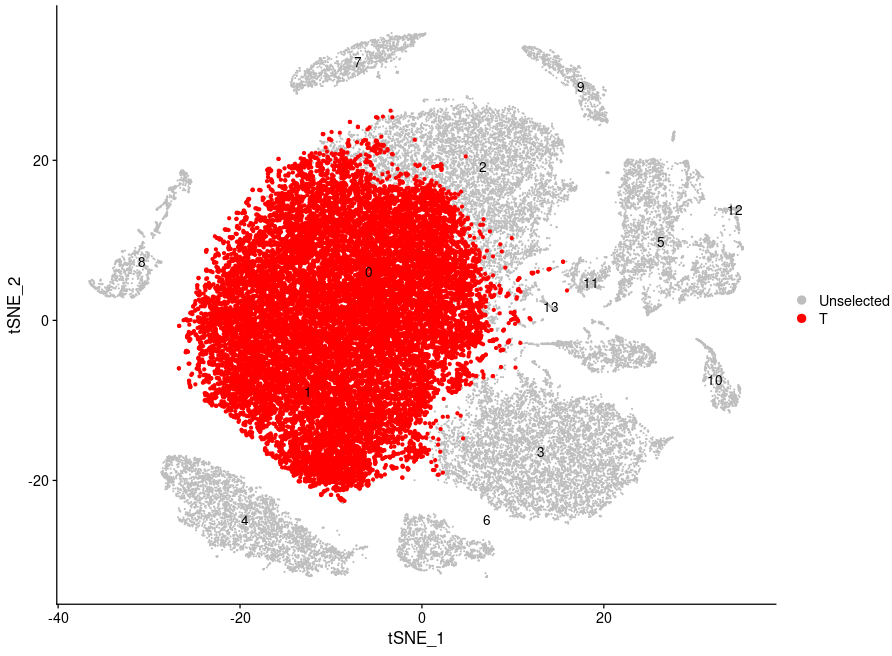
使用DimPlot函数展示T细胞("0","1")在tSNE图中的位置:
Idents(sce.all)
DimPlot(sce.all,label=T,
reduction = "tsne",pt.size = 0.2,
cells.highlight=list(
T=WhichCells(sce.all, idents = c("0","1"))
),
cols.highlight = c("red"),cols = "grey")
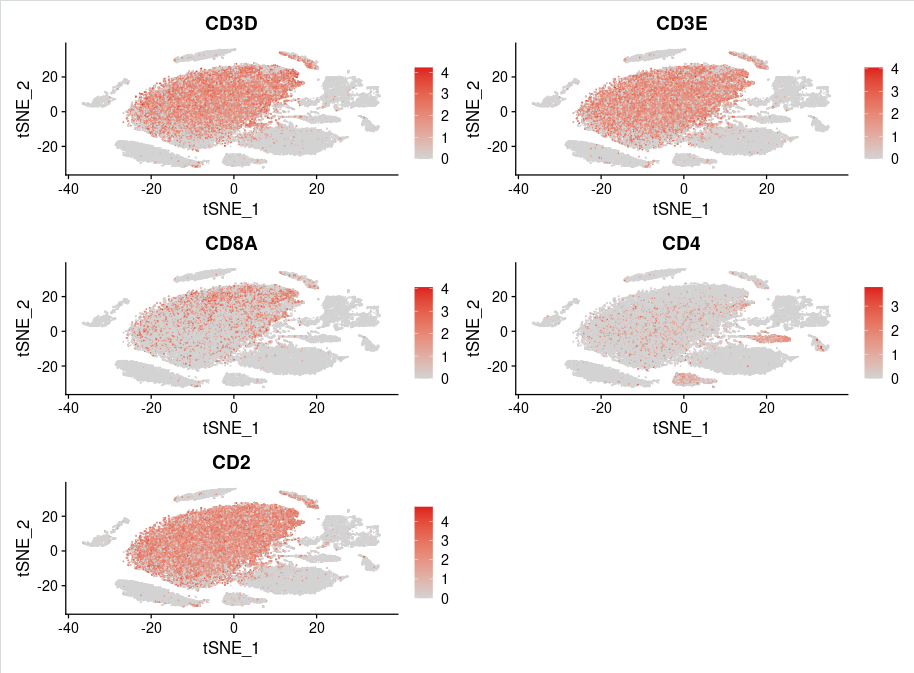
使用FeaturePlot函数展示T细胞maker基因('CD3D', 'CD3E', 'CD8A', 'CD4','CD2')的表达情况:
T_marker <- c('CD3D', 'CD3E', 'CD8A', 'CD4','CD2')
FeaturePlot(sce.all,features = T_marker,reduction = "tsne",cols = c("lightgrey" ,"#DE1F1F"),ncol=2,raster=FALSE)
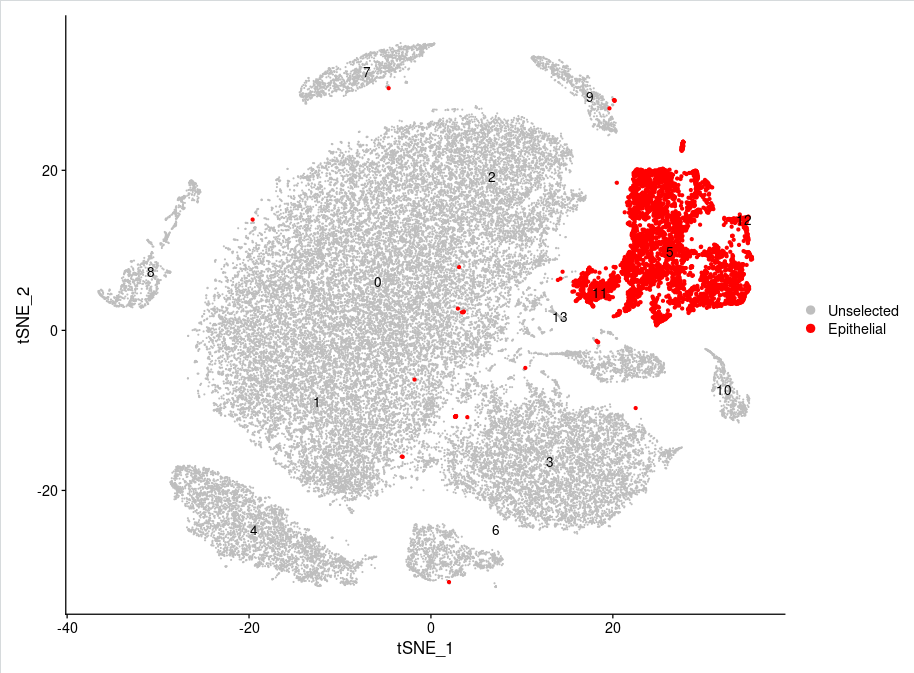
使用DimPlot函数展示上皮细胞("5","11","12")在tSNE图中的位置:
DimPlot(sce.all,label=T,
reduction = "tsne",pt.size = 0.2,
cells.highlight=list(
Epithelial=WhichCells(sce.all, idents = c("5","11","12"))
),
cols.highlight = c("red"),cols = "grey")
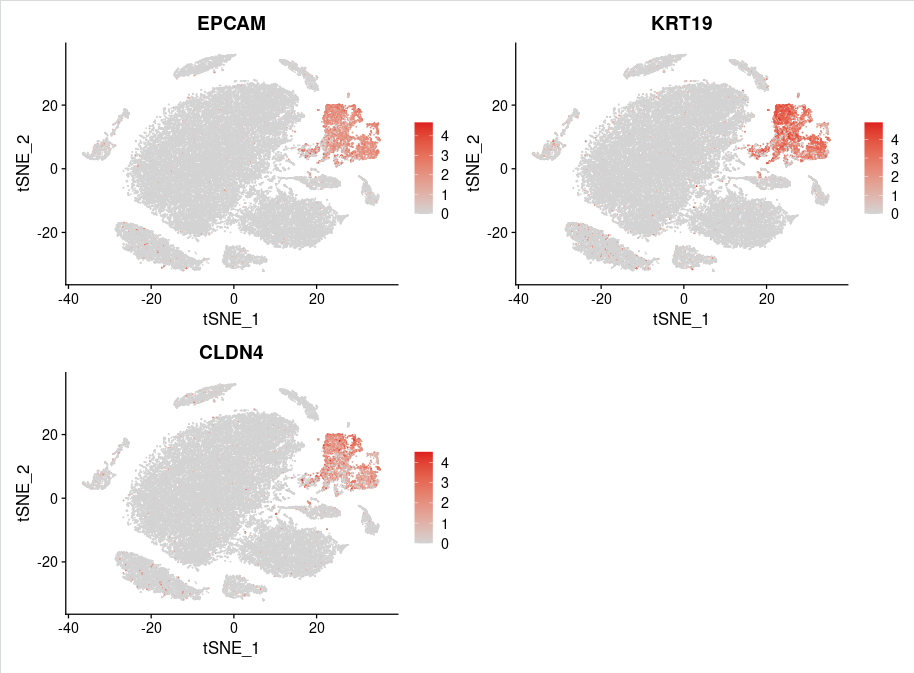
使用FeaturePlot函数展示上皮细胞maker基因('EPCAM','KRT19','CLDN4')的表达情况:
Epithelial_marker <- c('EPCAM','KRT19','CLDN4')
FeaturePlot(sce.all,features = Epithelial_marker,reduction = "tsne",cols = c("lightgrey" ,"#DE1F1F"),ncol=2,raster=FALSE)
FeaturePlot函数展示maker基因的表达也可以帮助我们检查注释准确与否,因此,我们可以批量绘制多个maker基因:
genes_to_check = c('EPCAM','KRT19','CLDN4', #上皮
'PECAM1' , 'CLO1A2', 'VWF', #基质
'CD3D', 'CD3E', 'CD8A', 'CD4','CD2', #T
'CDH5', 'PECAM1', 'VWF', #内皮
'LUM' , 'FGF7', 'MME', #成纤维
'AIF1', 'C1QC','C1QB','LYZ', #巨噬
'MKI67', 'STMN1', 'PCNA', #增殖
'CPA3' ,'CST3', 'KIT', 'TPSAB1','TPSB2',#肥大
'GOS2', 'S100A9','S100A8','CXCL8', #中性粒细胞
'KLRD1', 'GNLY', 'KLRF1','AREG', 'XCL2','HSPA6', #NK
'MS4A1','CD19', 'CD79A','IGHG1','MZB1', 'SDC1', #B
'CSF1R', 'CSF3R', 'CD68') #髓系
##批量画基因
FeaturePlot(sce.all, features =genes_to_check,
cols = c("lightgrey", 'red'),
reduction = "tsne",
ncol = 8 ) & NoLegend() & NoAxes() & theme(
panel.border = element_rect(color = "black", size = 1)
)
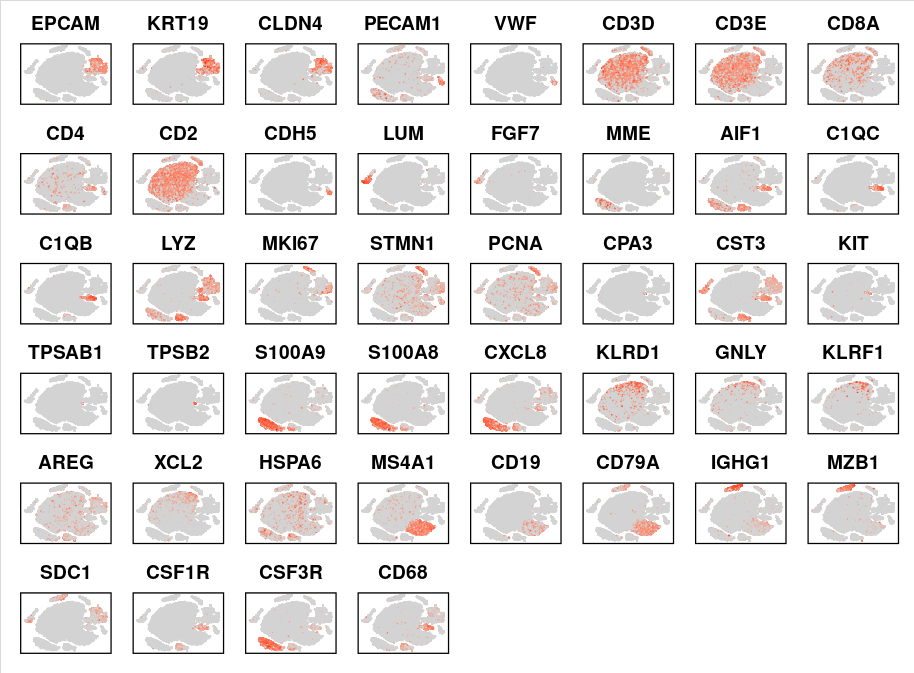
FeaturePlot除了可以展示单个基因,还可以把多个基因画在同一个图中。
Featureplot把两个基因画在同一个图中,看右上角可以发现黄色越深的地方两个基因叠加越多。这样可以看两个基因的共表达,是不是和免疫荧光图比较类似:
FeaturePlot(sce.all, features = c('S100A9','S100A8'),
reduction = "tsne",
cols = c("lightgrey", "green", "orange"),
blend=T,blend.threshold=0)

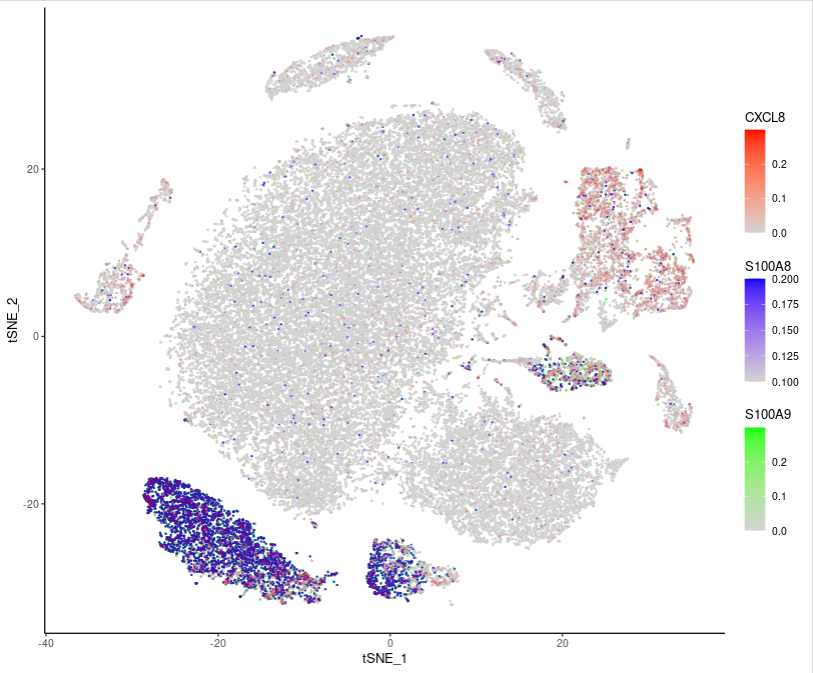
Featureplot还可以把三个基因画在同一个图中:
# 提取tsne坐标
tsne_df <- as.data.frame(sce.all@reductions$tsne@cell.embeddings)
tsne_df$cluster <- as.factor(sce.all$celltype)
head(tsne_df)
# 提取基因表达数据并与tsne坐标合并
gene_df <- as.data.frame(GetAssayData(object = sce.all, slot = "data")[c('S100A9','S100A8','CXCL8'), ])
library(ggnewscale)
merged_df <- merge(t(gene_df), tsne_df, by = 0, all = TRUE)
head(merged_df)
colnames(merged_df)
ggplot(merged_df, vars = c("tSNE_1", "tSNE_2", 'S100A9','S100A8','CXCL8'), aes(x = tSNE_1, y = tSNE_2, colour = S100A9)) +
geom_point(size=0.3, alpha=1) +
scale_colour_gradientn(colours = c("lightgrey", "green"), limits = c(0, 0.3), oob = scales::squish) +
new_scale_color() +
geom_point(aes(colour = S100A8), size=0.3, alpha=0.7) +
scale_colour_gradientn(colours = c("lightgrey", "blue"), limits = c(0.1, 0.2), oob = scales::squish) +
new_scale_color() +
geom_point(aes(colour = CXCL8), size=0.3, alpha=0.1) +
scale_colour_gradientn(colours = c("lightgrey", "red"), limits = c(0, 0.3), oob = scales::squish)+
theme_classic()
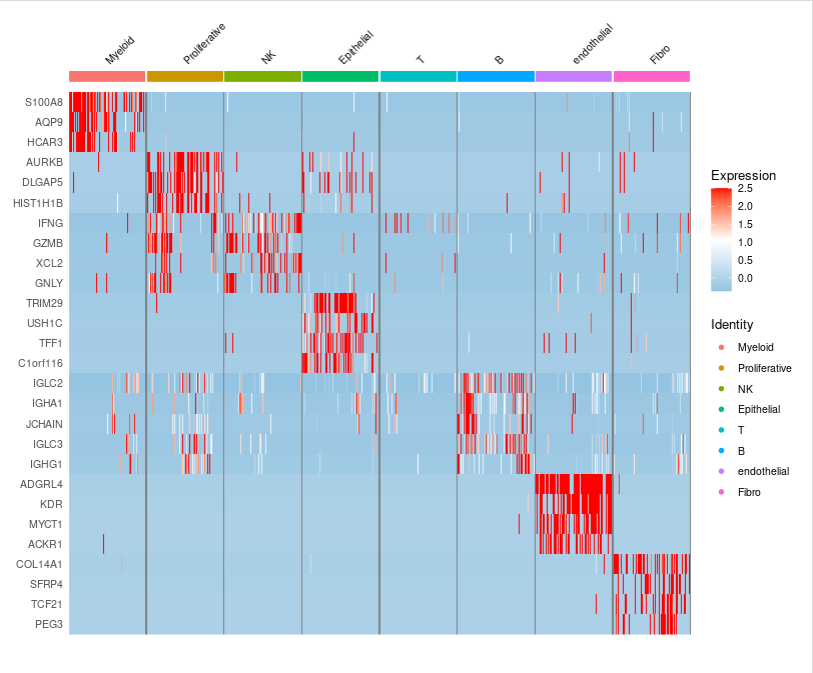
此外,我们还可以使用DoHeatmap函数绘制热图,展示每群细胞top基因:
Idents(sce.all)
Idents(sce.all) = sce.all$celltype
sce.markers <- FindAllMarkers(object = sce.all, only.pos = TRUE,
min.pct = 0.25,
thresh.use = 0.25)
write.csv(sce.markers,file='sce.markers.csv')
#sce.markers = read.csv('sce.markers.csv',row.names = 1)
library(dplyr)
top5 <- sce.markers %>% group_by(cluster) %>% top_n(5, avg_log2FC)
#为了防止数据量太大不好出图,这里在每个亚群提取出来100个
DoHeatmap(subset(sce.all,downsample=100),top5$gene,size=3)+scale_fill_gradientn(colors=c("#94C4E1","white","red"))
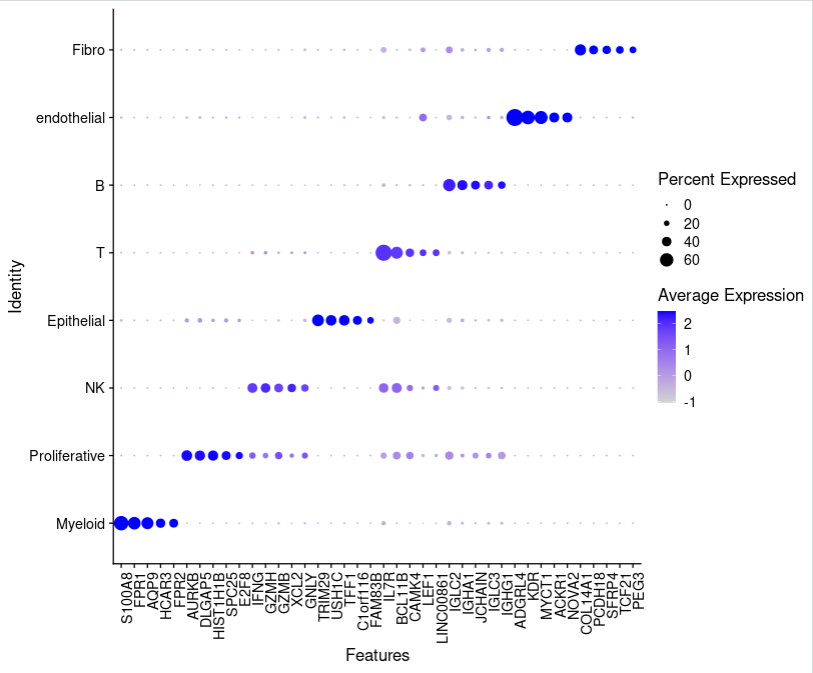
使用DotPlot函数绘制气泡图:
top5_dotplot <- DotPlot(sce.all, features = top5$gene)+
theme(axis.text.x = element_text(angle = 90, vjust = 1, hjust = 1))
top5_dotplot
结语
本期,我们使用了DimPlot、FeaturePlot、DoHeatmap、DotPlot等多种函数对细胞群和基因进行了可视化。下一期,我们将在此基础上,绘制饼图、堆积柱状图、箱线图、气泡图等,比较不同分组之间细胞比例差异。干货满满,欢迎大家持续追更,谢谢!
第五集:细胞比例
1.背景
在细胞分群命名完成之后,我们可以比较不同样本和分组之间细胞比例的差异。细胞发生癌变、肿瘤细胞转移、药物治疗等刺激因素,都会导致肿瘤微环境中细胞类型发生改变。不同类型的细胞执行不同的生物学功能,通过计算细胞比例,我们可以评估细胞类型的组内及组间分布情况。
2.可视化
在这里,我们将依次绘制饼图、堆积柱状图、气泡图。
2.1 饼图
饼图可以直观展示组内各细胞比例的差异。
首先加载R包:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(dplyr)
library(plotrix)
library(ggsci)
library(celldex)
library(singleseqgset)
library(devtools)
创建工作目录,并读取细胞分群注释后数据:
getwd()
setwd()
dir.create("5-prop")
setwd('5-prop/')
sce.all=readRDS( "../3-Celltype/sce_celltype.rds")
sce.all
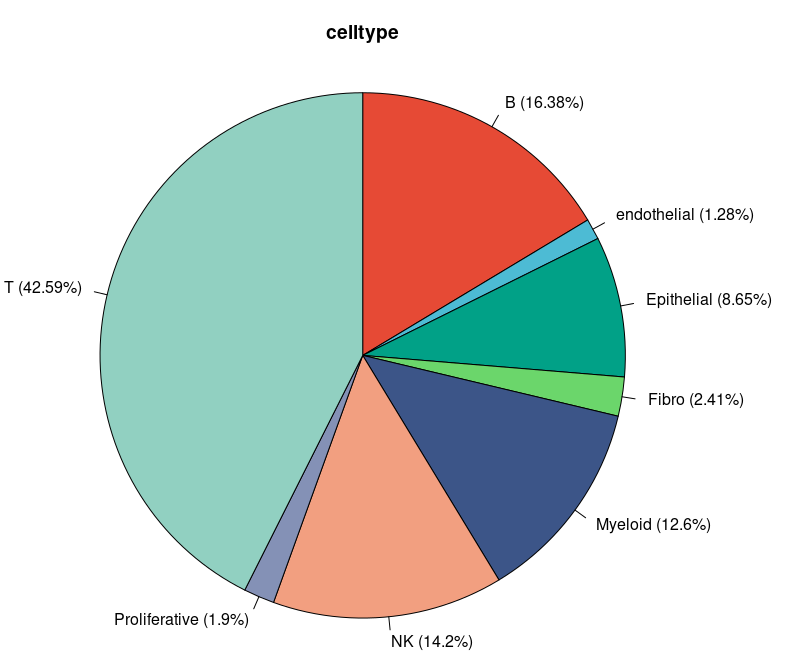
绘制饼图:
head(sce.all@meta.data)
table(sce.all$celltype)
mynames <- table(sce.all$celltype) %>% names()
myratio <- table(sce.all$celltype) %>% as.numeric()
pielabel <- paste0(mynames," (", round(myratio/sum(myratio)*100,2), "%)")
cols <-c('#E64A35','#4DBBD4' ,'#01A187','#6BD66B','#3C5588' ,'#F29F80' ,
'#8491B6','#91D0C1','#7F5F48','#AF9E85','#4F4FFF','#CE3D33',
'#739B57','#EFE685','#446983','#BB6239','#5DB1DC','#7F2268','#800202','#D8D8CD'
)
pie(myratio, labels=pielabel,
radius = 1.0,clockwise=T,
main = "celltype",col = cols)
绘制3D饼图:
pie3D(myratio,labels = pielabel,explode = 0.1,
main = "Cell Proption",
height = 0.3,
labelcex = 1)

pie函数参数说明:
x: 数值向量,表示每个扇形的面积。
labels: 字符型向量,表示各扇形面积标签。
edges: 这个参数用处不大,指的是多边形的边数(圆的轮廓类似很多边的多边形)。
radius: 饼图的半径。
main: 饼图的标题。
clockwise: 是一个逻辑值,用来指示饼图各个切片是否按顺时针做出分割。
angle: 设置底纹的斜率。
density: 底纹的密度。默认值为 NULL。
col: 是表示每个扇形的颜色,相当于调色板。
2.2 堆积柱状图
堆积柱状图除了可以展示组内差异,还可以直观展示组间细胞比例差异。
绘制堆积柱状图前,首先整理图形输入数据:
library(tidyr)
library(reshape2)
tb=table(sce.all$tissue, sce.all$celltype)
head(tb)
library (gplots)
balloonplot(tb)
bar_data <- as.data.frame(tb)
bar_per <- bar_data %>%
group_by(Var1) %>%
mutate(sum(Freq)) %>%
mutate(percent = Freq / `sum(Freq)`)
head(bar_per)
#write.csv(bar_per,file = "celltype_by_group_percent.csv")
col =c("#3176B7","#F78000","#3FA116","#CE2820","#9265C1",
"#885649","#DD76C5","#BBBE00","#41BED1")
colnames(bar_per)
可视化:
library(ggthemes)
p1 = ggplot(bar_per, aes(x = percent, y = Var1)) +
geom_bar(aes(fill = Var2) , stat = "identity") + coord_flip() +
theme(axis.ticks = element_line(linetype = "blank"),
legend.position = "top",
panel.grid.minor = element_line(colour = NA,linetype = "blank"),
panel.background = element_rect(fill = NA),
plot.background = element_rect(colour = NA)) +
labs(y = " ", fill = NULL)+labs(x = 'Relative proportion(%)')+
scale_fill_manual(values=col)+
theme_few()+
theme(plot.title = element_text(size=12,hjust=0.5))
p1
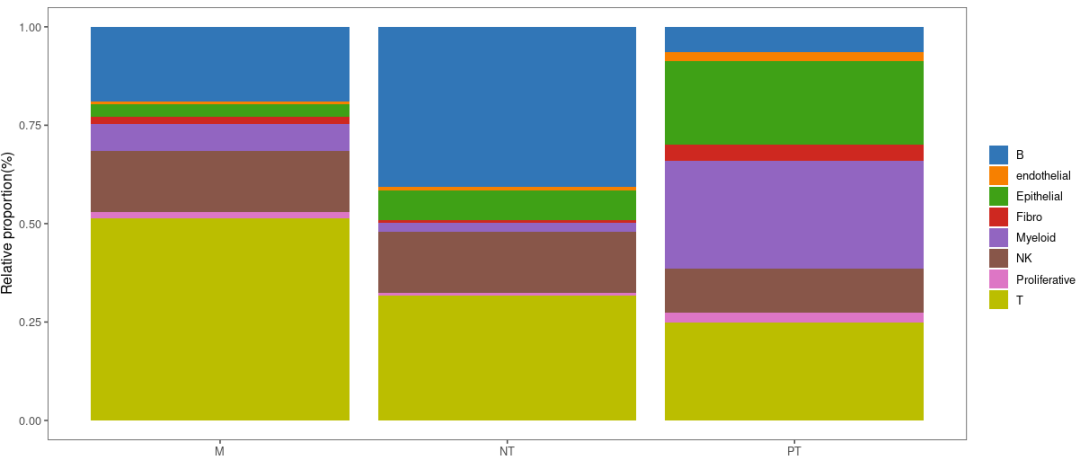
上图是按样本类型(M、NT、PT)分组比较的,我们也可以按患者进行比较:
bar_data <- as.data.frame(tb)
bar_per <- bar_data %>%
group_by(Var1) %>%
mutate(sum(Freq)) %>%
mutate(percent = Freq / `sum(Freq)`)
head(bar_per)
#write.csv(bar_per,file = "celltype_by_group_percent.csv")
col =c("#3176B7","#F78000","#3FA116","#CE2820","#9265C1",
"#885649","#DD76C5","#BBBE00","#41BED1")
colnames(bar_per)
library(ggthemes)
p2 = ggplot(bar_per, aes(x = percent, y = Var1)) +
geom_bar(aes(fill = Var2) , stat = "identity") + coord_flip() +
theme(axis.ticks = element_line(linetype = "blank"),
legend.position = "top",
panel.grid.minor = element_line(colour = NA,linetype = "blank"),
panel.background = element_rect(fill = NA),
plot.background = element_rect(colour = NA)) +
labs(y = " ", fill = NULL)+labs(x = 'Relative proportion(%)')+
scale_fill_manual(values=col)+
theme_few()+
theme(plot.title = element_text(size=12,hjust=0.5)) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
p2
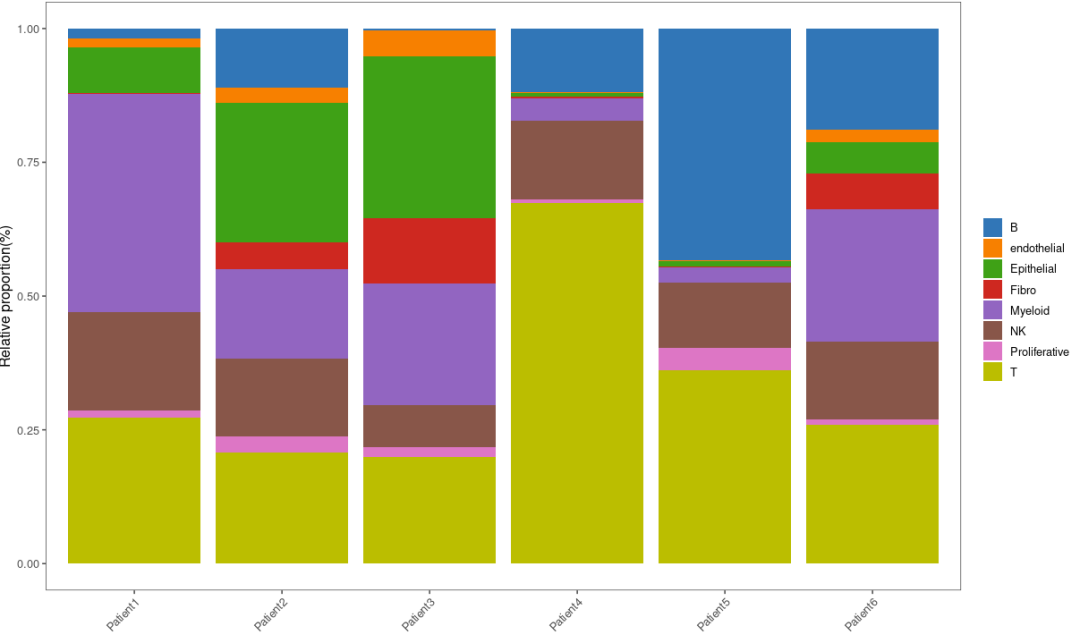
X、Y轴转换一下:
p3 <- ggplot(bar_per, aes(y = percent, x = Var1)) +
geom_bar(aes(fill = Var2) , stat = "identity") + coord_flip() +
theme(axis.ticks = element_line(linetype = "blank"),
legend.position = "top",
panel.grid.minor = element_line(colour = NA,linetype = "blank"),
panel.background = element_rect(fill = NA),
plot.background = element_rect(colour = NA)) +
labs(y = " ", fill = NULL)+labs(x = 'Relative proportion(%)')+
scale_fill_manual(values=col)+
theme_few()+
theme(plot.title = element_text(size=12,hjust=0.5)) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
p3
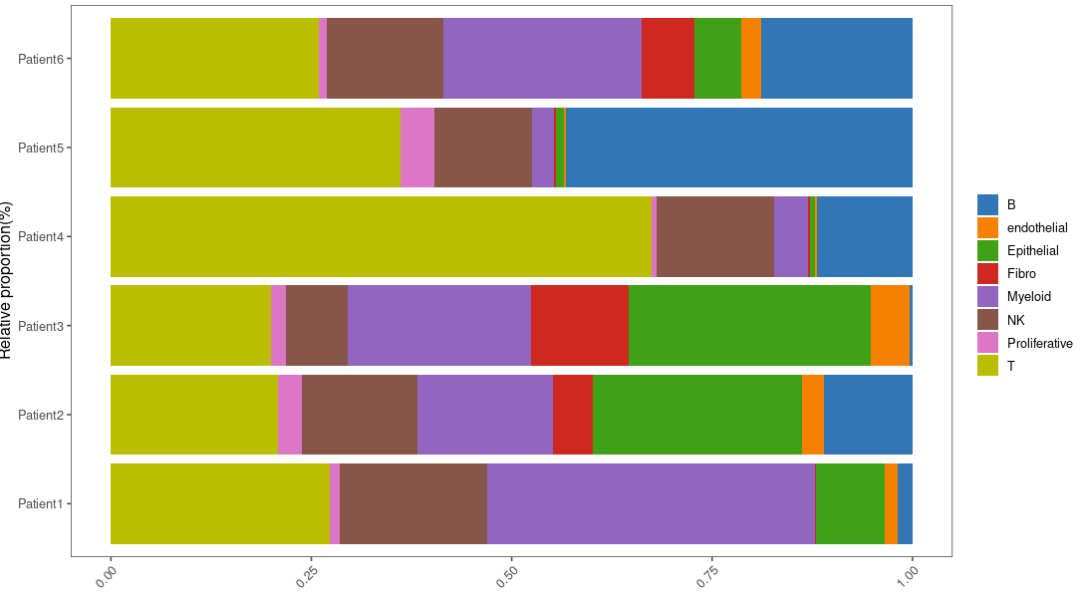
ggplot函数参数说明:
原文链接:https://blog.csdn.net/a11113112/article/details/135091751
data:要用于绘图的数据集。
mapping 或 aes:美学映射,用于将数据变量映射到图形属性,例如 x、y、color、size、shape 等。
geom:几何对象,确定图形类型,比如 geom_point()(散点图)、geom_line()(折线图)、geom_bar()(条形图)等。
x、y:指定 x 轴和 y 轴的数据变量。
color、fill、shape、size:用于指定颜色、填充、形状和大小的变量。
alpha:指定颜色透明度。
group:指定分组变量。
facet:面板分组,允许在一个图中绘制多个小图(facet_wrap() 或 facet_grid())。
theme:用于设置图形的主题样式。
labs:用于设置 x、y 轴标签和图例标签的文本。
scale:用于调整比例尺和美学属性的尺度。
coord:坐标系变换,例如 coord_flip() 可以交换 x 和 y 轴。
position:用于调整图形中元素的位置,例如 position_dodge() 用于避免重叠的条形或点。
xlim、ylim:控制 x 轴和 y 轴的绘图范围。
coord_cartesian():类似于 xlim 和 ylim,但不会删除超出范围的数据点。
labs():设置图形的标题、坐标轴标签和图例标题。
ggtitle():设置图形的主标题。
theme():调整图形的外观和布局,如背景、网格线、标签样式等。
guides():控制图例的外观,如标题、标签和图例键的位置和样式。
scale_x_continuous()、scale_y_continuous():调整 x 轴和 y 轴的连续变量的比例尺和标签。
.......
2.3 气泡图
气泡图则可以展示每组样本不同类型细胞的具体数量。
绘制气泡图:
unique(sce.all@meta.data$patient)
tb=table(sce.all$patient, sce.all$celltype)
head(tb)
library (gplots)
balloonplot(tb)

可以调整字体大小:
balloonplot(tb, text.size=0.8,label.size=0.8)
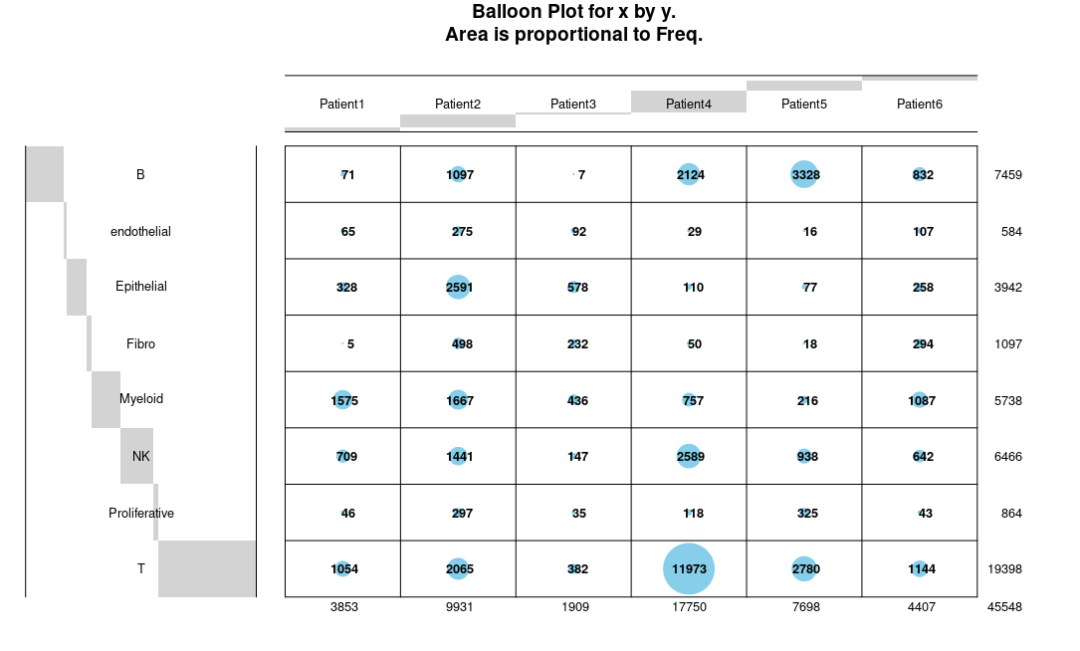
还可以调整X/Y标签的角度:
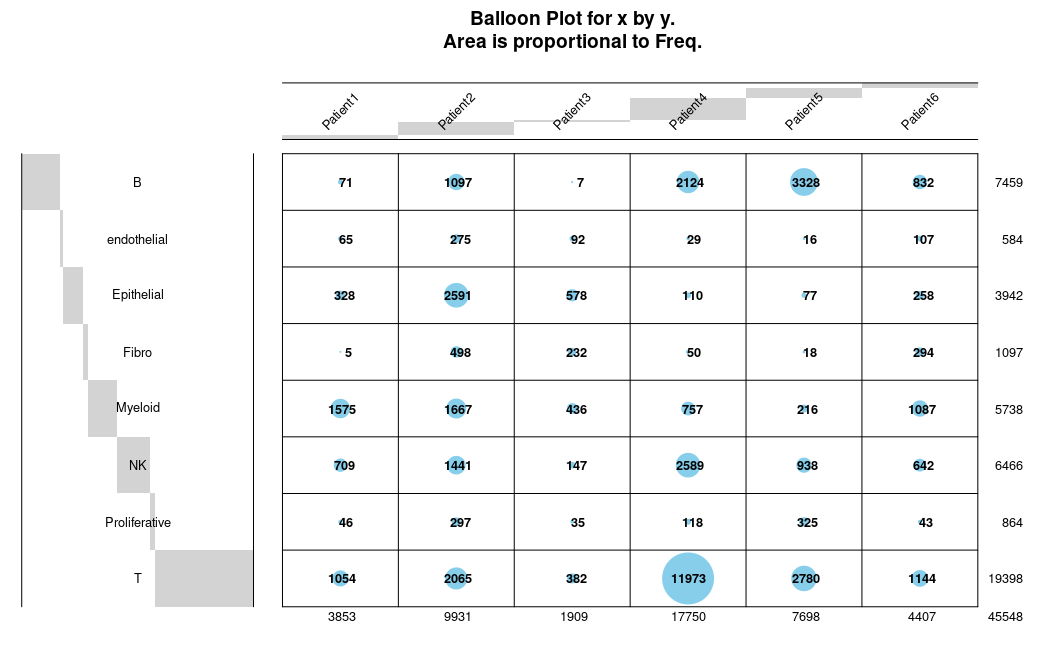
以及标签字体的颜色:
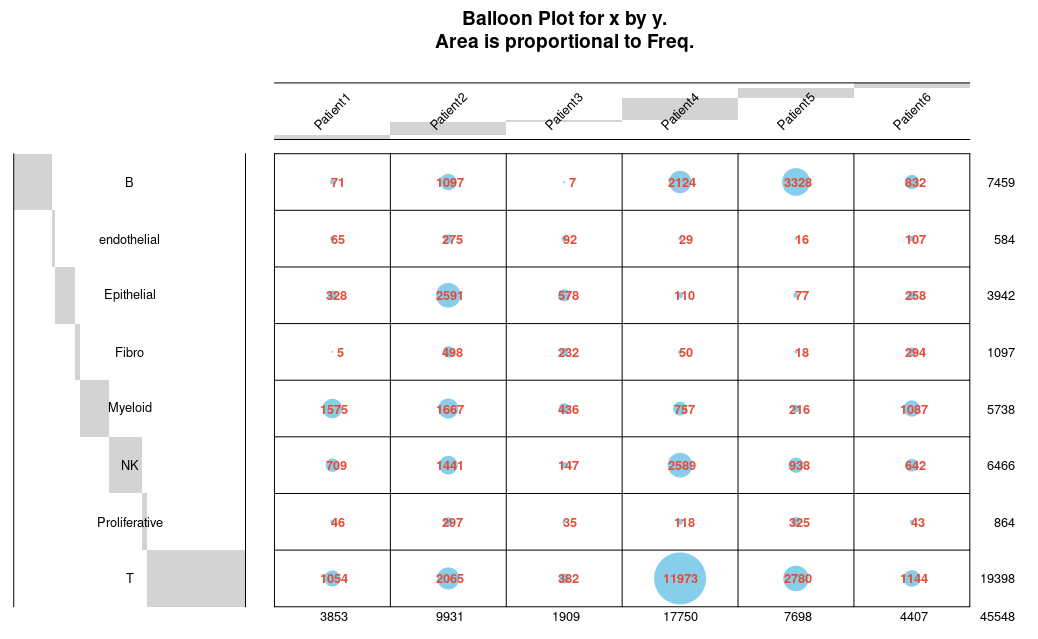
balloonplot函数参数说明:
x : 一个表对象,或一个向量或几个类别向量的列表,其中包含打印矩阵的第一个(x)边距的分组变量。
y : 矢量或矢量列表,用于对绘制矩阵的第二(y)维变量进行分组。z : 打印矩阵中点大小的值向量。
xlab : x维度的文本标签。这将显示在X轴和绘图标题中。
ylab : y标注的文本标签。这将显示在坐标轴和绘图标题中。
zlab : 点大小的文本标签。这将包含在地块标题中。
dotsize : 最大点大小。可能需要为不同的打印设备和布局调整此值。
dotchar : 用于点的绘图符号或字符。有关符号代码的points函数,请参见帮助页。
dotcolor : 指定绘图点颜色的标量或向量。
text.size, text.color : 行和列标题的字符大小和颜色
.......
结语
本期,我们绘制饼图、堆积柱状图、箱线图、气泡图等,比较不同分组之间细胞比例差异。下一期,我们将利用胃癌bulk数据(TCGA-STAD)协助区分正常上皮和恶性上皮,其中还将涵盖差异分析、富集分析、KM生存分析等bulk RNA-seq数据的常规分析。干货满满,欢迎大家持续追更,谢谢!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-07-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号