基于Python语言使用DESeq2方法进行bulk-RNAseq的差异表达分析

基于Python语言使用DESeq2方法进行bulk-RNAseq的差异表达分析

DoubleHelix
发布于 2024-04-15 10:34:20
发布于 2024-04-15 10:34:20
import pandas as pd
import numpy as np
import anndata as ad
import math
import seaborn as sns
import matplotlib.pyplot as plt
from pydeseq2.dds import DeseqDataSet
from pydeseq2.ds import DeseqStats
# from pydeseq2.utils import load_example_data读入count矩阵文件:
count_file = "data/count.csv"
counts_df = pd.read_csv(count_file,index_col=0).T
counts_df.head().iloc[:,range(10)]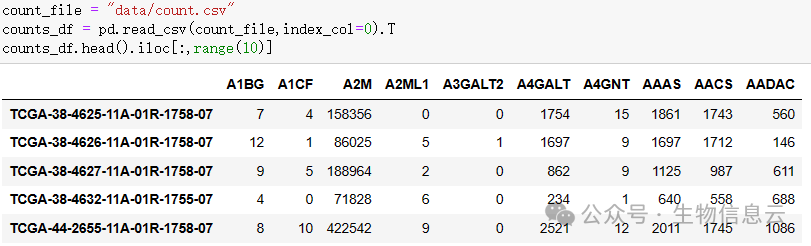
注意:读入的数据进行转置,是因为使用pydeseq2包进行分析时,count矩阵需要的是行为样本,列为基因名称,和R语言中的DESeq2包刚好相反。
读入样本信息文件:
condition_file = "data/matedata.csv"
condition_df = pd.read_csv(condition_file, index_col=0)
condition_df.head()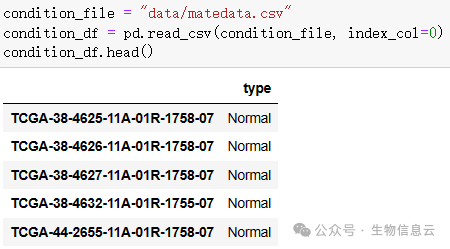
构建DeseqDataSet 对象,并进行差异分析:
# 构建DeseqDataSet 对象
dds = DeseqDataSet(counts = counts_df, clinical = condition_df, design_factors = "type")
# 离散度和log fold-change评估.
dds.deseq2()执行统计检验:
# 执行统计分析并返回结果
res = DeseqStats(dds)
res.summary()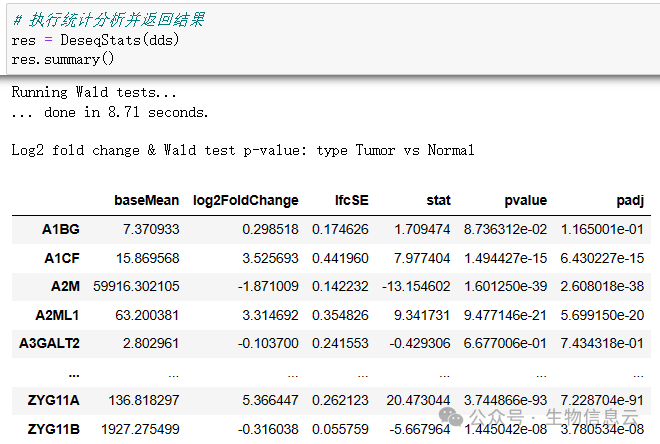
提取结果:
res_df = res.results_df
res_df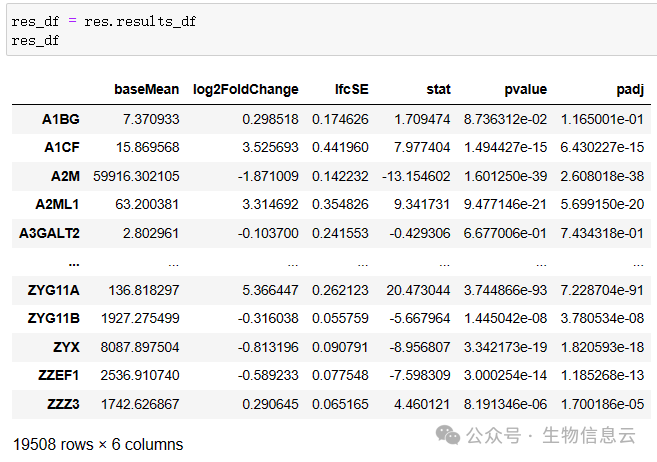
筛选显著性差异表达的基因:
# 对padj取-log10对数
res_df['-logpadj'] = -res_df.padj.apply(math.log10)
# 尝试写循环筛选上下调基因分类赋值给 "up" 和 "down" 和 "nosig" 加入pvalue条件
###loc函数:通过行索引 "Index" 中的具体值来取行数据(如取"Index"为"A"的行)
res_df.loc[(res_df.log2FoldChange>2)&(res_df.padj<0.05),'type']='up'
res_df.loc[(res_df.log2FoldChange<-2)&(res_df.padj<0.05),'type']='down'
res_df.loc[(abs(res_df.log2FoldChange)<=2)|(res_df.padj>=0.05),'type']='nosig'
res_df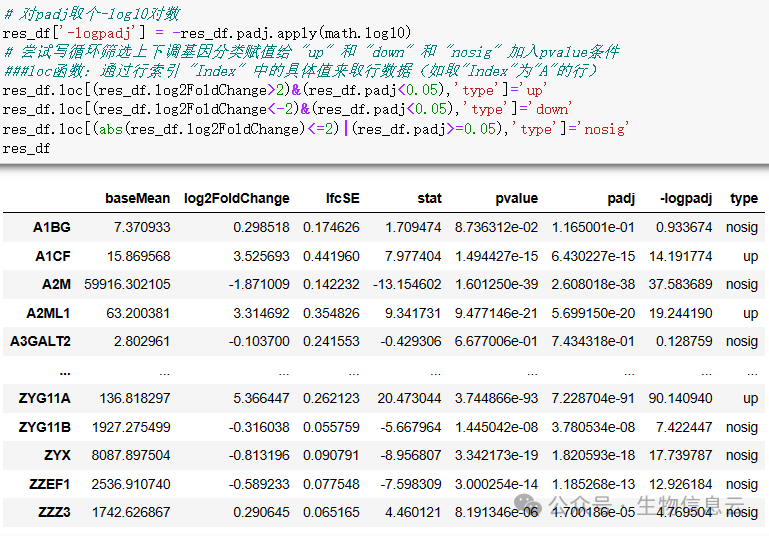
res_df.type.value_counts()
火山图可视化:
# 先设置一下颜色
colors = ["#808080","#DC143C","#00008B"]
sns.set_palette(sns.color_palette(colors))
# 绘图
ax=sns.scatterplot(x='log2FoldChange', y='-logpadj',data = res_df,
hue='type',#颜色映射
edgecolor = None,#点边界颜色
s=8,#点大小
)
# 标签
ax.set_title("Vocalno")
ax.set_xlabel("log2FoldChange")
ax.set_ylabel("-log10(padj)")
#移动图例位置
ax.legend(loc='center right', bbox_to_anchor=(0.95,0.76), ncol=1)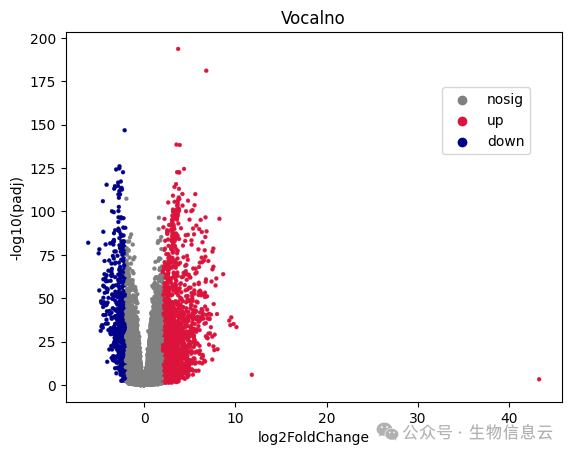
保存结果:
res_df.to_csv("python_deseq2.csv")本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-04-10,如有侵权请联系 cloudcommunity@tencent.com 删除
本文分享自 MedBioInfoCloud 微信公众号,前往查看
如有侵权,请联系 cloudcommunity@tencent.com 删除。
本文参与 腾讯云自媒体同步曝光计划 ,欢迎热爱写作的你一起参与!
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号