美化clusterProfiler富集分析结果:enrichplot包中的cnetplot
原创
美化clusterProfiler富集分析结果:enrichplot包中的cnetplot
原创
生信小博士
发布于 2024-03-30 18:26:54
发布于 2024-03-30 18:26:54
写在开头
相信大家对富集分析都很熟悉,但是对富集分析结果的美化却永无止境。
今天我们介绍Y叔系列中enrichplot包的cnetplot函数。
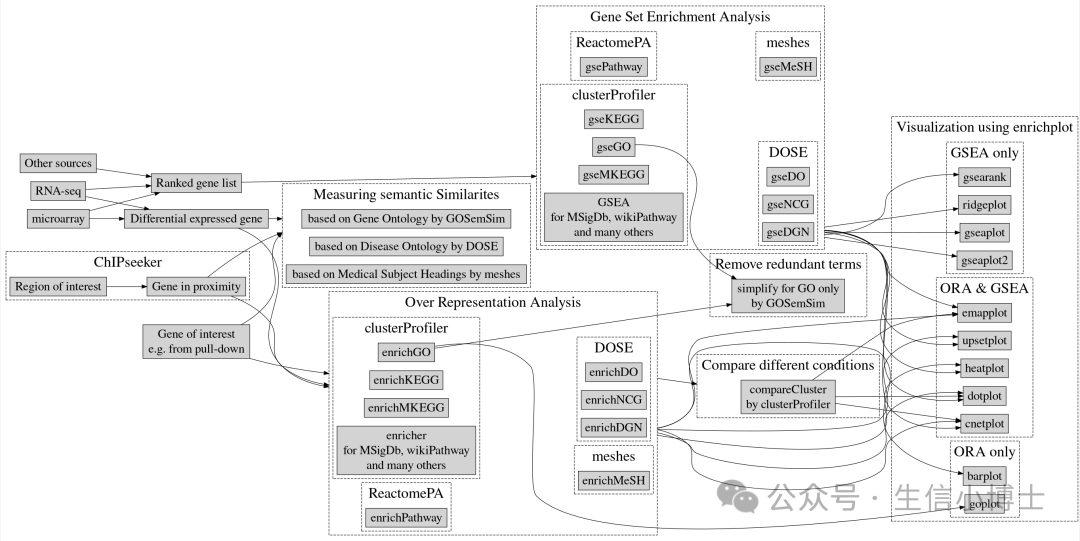
cnetplot函数介绍
Y叔为啥要开发cnetplot函数来画图呢,需要我们先理解一个概念:基因-概念网络 (Gene-Concept Network) GCN
- barplot() 和 dotplot() 函数通常只显示最重要的或选定的富集术语,这些术语代表基因表达或功能上的富集。然而,使用者可能想知道哪些基因参与了这些关键的富集术语。
- 为了更全面地考虑基因的生物学复杂性,例如一个基因可能属于多个功能类别并可能存在表达量的变化,研究人员开发了 cnetplot() 函数来提取基因与相关概念之间的复杂关联信息。
- cnetplot() 函数将基因和生物学概念(例如 Gene Ontology 条目或 KEGG 通路)之间的关联描绘成一个网络,从而直观地展示基因与功能术语之间的关系。
- 除了标准的富集分析结果,cnetplot() 函数也支持基因集富集分析 (GSEA) 的结果展示,并仅显示核心富集基因。
cnetplot函数所需要的输入内容:
library(DOSE)data(geneList);head(geneList)de <- names(geneList)[abs(geneList) > 2]edo <- enrichDGN(de);head(edo)
cnetplot函数小试牛刀,先画barplot看看
barplot,此函数只能对接enrichResult对象,所以GSEA的结果它是画不出来的 ,barplot用于展示最重要的或者你感兴趣的条目的富集结果,比如富集到的基因个数、条目名字,P值等信息。
library(enrichplot)barplot(edo, showCategory=20) ;head(edo)
library(enrichplot)
p1 <- barplot(ora_res, showCategory=10 # 展示多少条目 ,x = "Count" # X轴展示那个变量,默认Count,也可以是GeneRatio ,label_format = 30 # 默认对名字超过30个字符的进行折叠 ,font.size = 12 # 字体大小 ,title = "Bar plot for ORA" )
p2 <- barplot(ora_res, showCategory=10 ,x = "GeneRatio" )
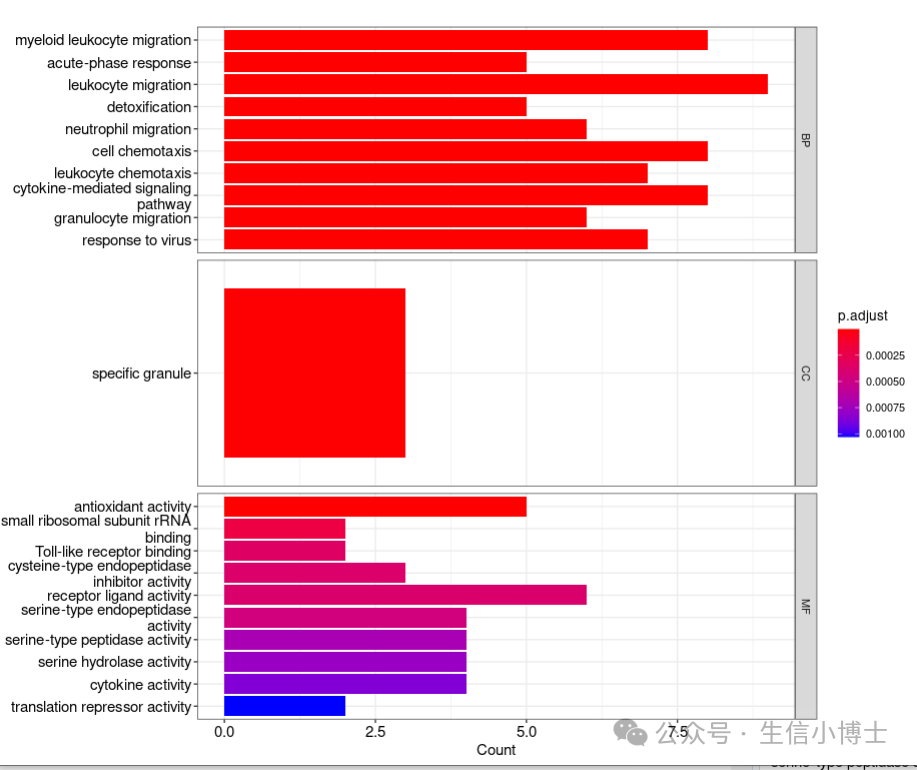
cowplot::plot_grid(p1,p2)barplot(ora_res, showCategory=10 ,split = "ONTOLOGY" # 分面,GO ORA特有 ) + facet_grid(ONTOLOGY~., scale="free") # ggplot2的分面语法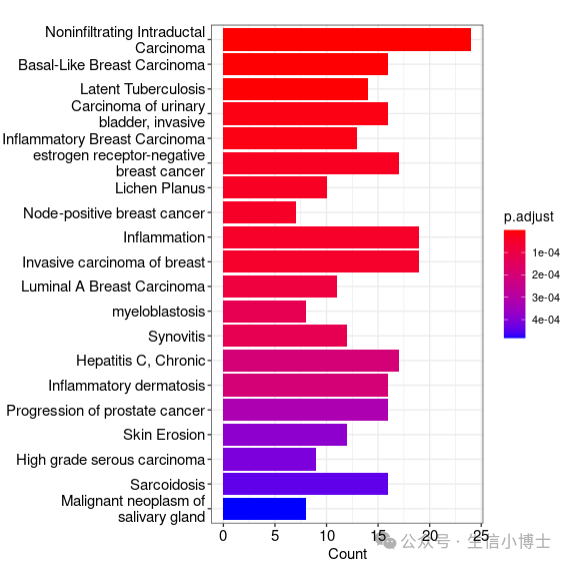
cnetplot可以给基因加上foldchange信息,让图更好看
#3 readable-- edox <- setReadable(edo, 'org.Hs.eg.db', 'ENTREZID')
cnetplot(edox, foldChange=geneList) ## categorySize can be scaled by 'pvalue' or 'geneNum'
cnetplot(edox, categorySize="pvalue", foldChange=geneList)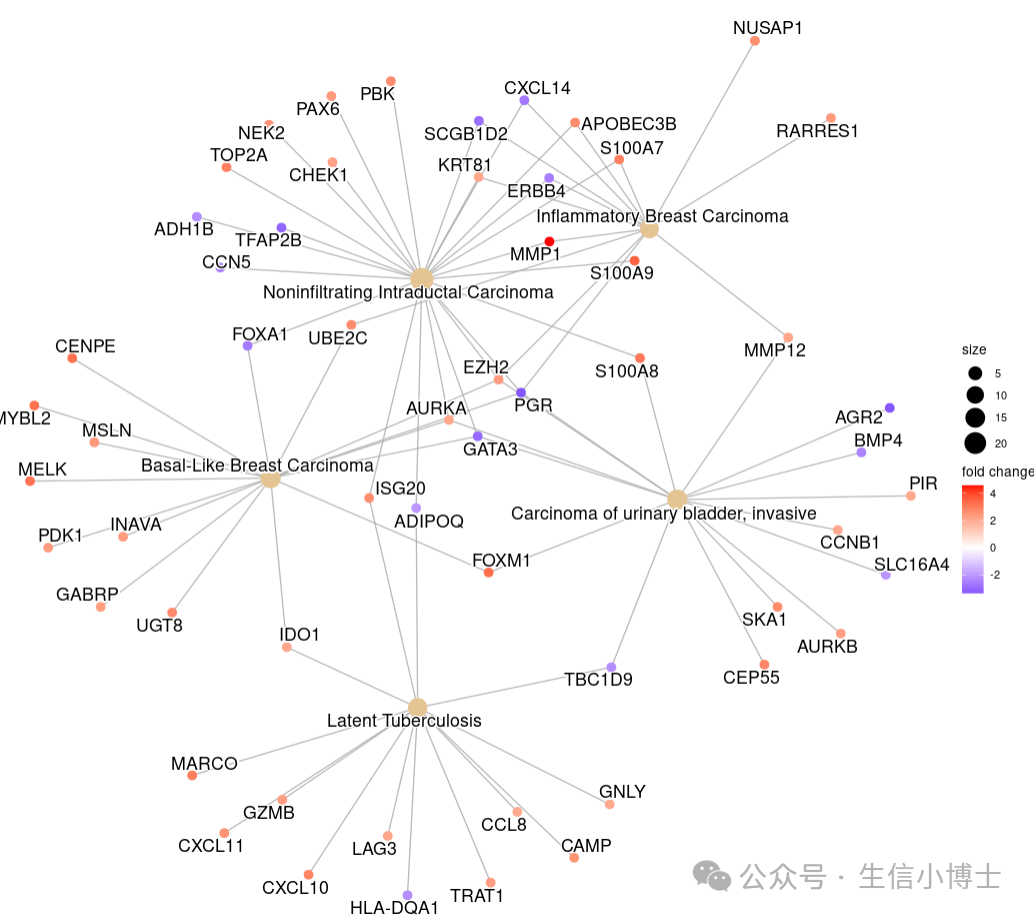
下面这张有点像弦图
cnetplot(edox, foldChange=geneList, circular = TRUE, colorEdge = TRUE)
富集分析结果也可画成热图形式
p1 <- cnetplot(edox, node_label="category")
p2 <- cnetplot(edox, node_label="gene")
p3 <- cnetplot(edox, node_label="all")
p4 <- cnetplot(edox, node_label="none")
cowplot::plot_grid(p1, p2, p3, p4, ncol=2, labels=LETTERS[1:4])
heatplot(edox, foldChange=geneList)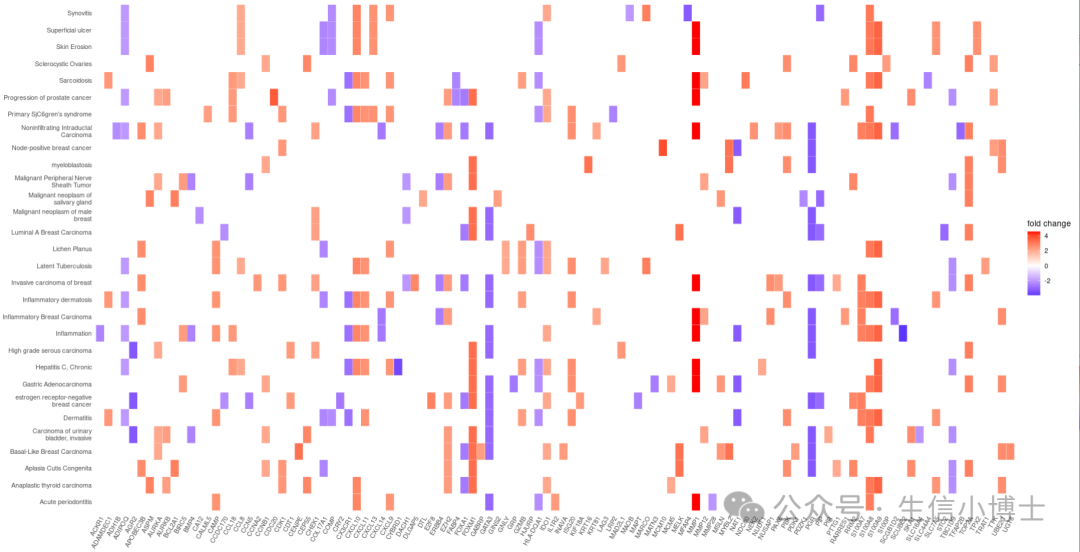
下面这段代码有助于理解cnetplot画图原理
#8理解cnetplot画图原理 ------ set.seed(123) x <- list(A = letters[1:10], B=letters[5:12], C=letters[sample(1:26, 15)]);x p1 <- cnetplot(x) set.seed(123) d <- setNames(rnorm(26), letters);d p2 <- cnetplot(x, foldChange=d) + scale_color_gradient2(name='associated data', low='darkgreen', high='firebrick');p2 cowplot::plot_grid(p1, p2, ncol=2, labels=LETTERS[1:2])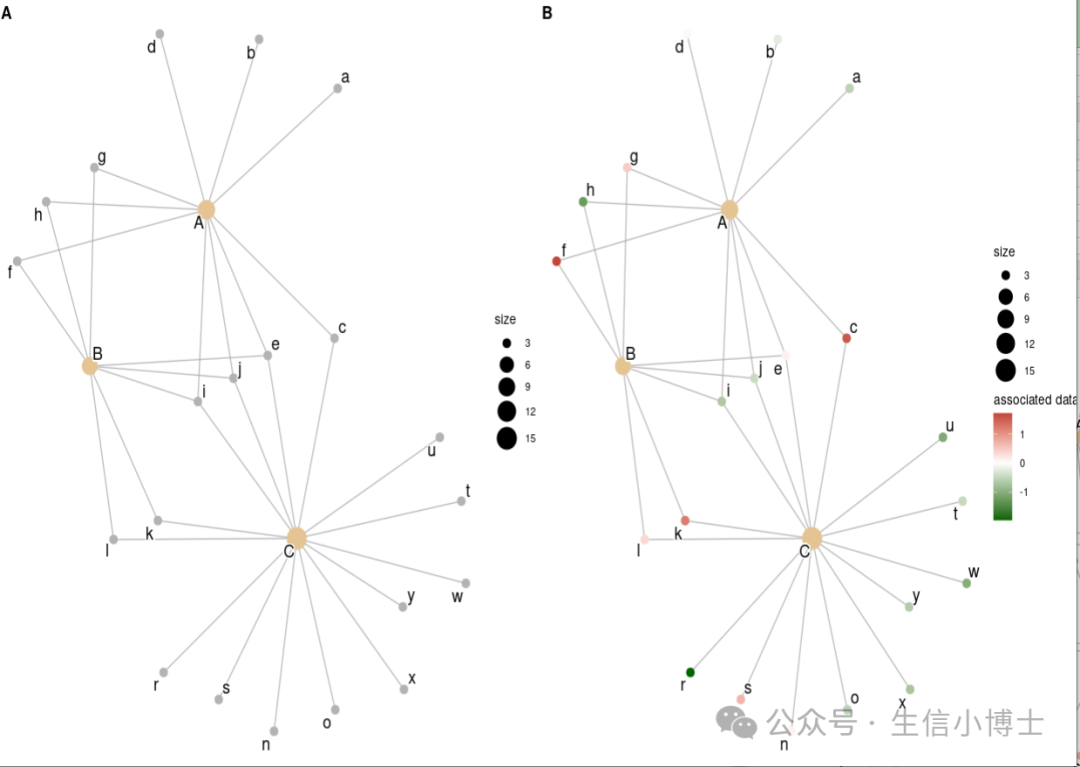
展示kegg图
library("pathview")
hsa04110 <- pathview(gene.data = geneList, pathway.id = "hsa04110",
species = "hsa",
limit = list(gene=max(abs(geneList)), cpd=1))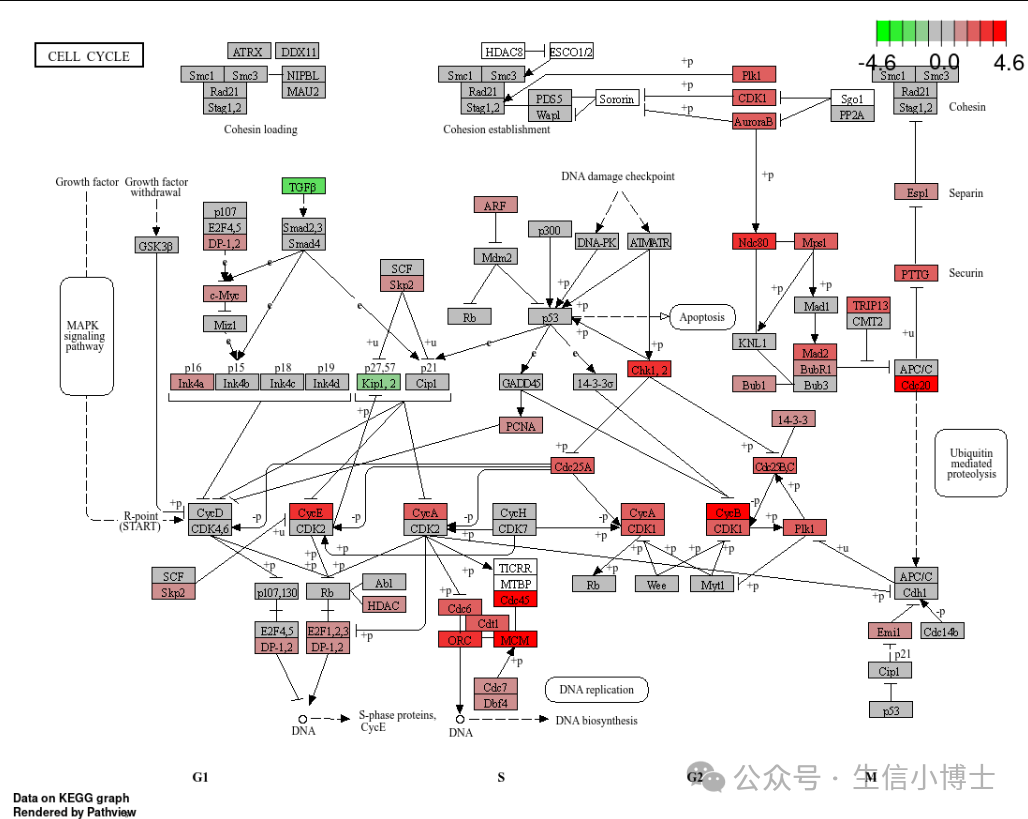
最后,我们复习一下cnetplot函数的用法:
函数简介:
cnetplot 函数用于可视化基因与生物学概念之间关联的函数,它可以将基因和生物学概念(例如 Gene Ontology 条目或 KEGG 通路)之间的关联描绘成一个网络,从而直观地展示基因与功能术语之间的关系。
参数:
- x:包含富集分析结果的 R 对象。
- foldChange:基因的表达量变化值。
- colorEdge:是否根据富集术语对边进行着色。
- category:类别节点的颜色。
- color_gene:基因节点的颜色。
- node_label="category" #节点标签 “category”, “gene”, “all” and “none” cex_label_category = 1.2 #节点标签字体大小
输出结果:
cnetplot 函数输出一个基因-概念网络,其中:
- 节点代表基因或生物学概念。
- 边代表基因与生物学概念之间的关联。
- 节点的颜色代表基因的表达量变化值或生物学概念的类型。
## convert gene ID to Symboledox <- setReadable(edo, 'org.Hs.eg.db', 'ENTREZID')
p1 <- cnetplot(edox, foldChange=geneList)## categorySize can be scaled by 'pvalue' or 'geneNum'
p2 <- cnetplot(edox, categorySize="pvalue", foldChange=geneList)
p3 <- cnetplot(edox, foldChange=geneList, circular = TRUE, colorEdge = TRUE)
cowplot::plot_grid(p1, p2, p3, ncol=3, labels=LETTERS[1:3], rel_widths=c(.8, .8, 1.2))
参考:https://yulab-smu.top/biomedical-knowledge-mining-book/index.html#可视化:https://yulab-smu.top/biomedical-knowledge-mining-book/enrichplot.html
微信公众号:生信小博士
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号