harmony包整合多批次单细胞数据以去除批次效应(batch effect)

harmony包整合多批次单细胞数据以去除批次效应(batch effect)

DoubleHelix
发布于 2023-12-26 11:48:19
发布于 2023-12-26 11:48:19
单细胞分析的前期教程:
单细胞专题 | 1.单细胞测序(10×genomics技术)的原理 单细胞专题 | 2.如何开始单细胞RNASeq数据分析 单细胞专题 | 3.单细胞转录组的上游分析-从BCL到FASTQ 单细胞专题 | 4.单细胞转录组的上游分析-从SRA到FASTQ 单细胞专题 | 5.单细胞转录组的上游分析-从FASTQ到count矩阵 单细胞专题 | 6.单细胞下游分析——不同类型的数据读入 单细胞专题 | 7.单细胞下游分析——常规分析流程案例一 单细胞专题 | 8.单细胞类型注释之SingleR包详解 单细胞专题 | 9.如何人工注释单细胞类群? 单细胞专题 | 10.细胞周期分析 单细胞专题 | 11.最新版cellphoneDB细胞通讯分析
单细胞专题 | 12.cellChat细胞通讯代码案例
1.1 方法文献
Korsunsky, I., Millard, N., Fan, J. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). https://doi.org/10.1038/s41592-019-0619-0
1.2 使用案例
https://github.com/immunogenomics/harmony
(1) 加载案例数据,并构建Seurat对象
## Source required libraries
## install.packages("harmony")
library(data.table)
library(tidyverse)
library(ggthemes)
library(ggrepel)
library(harmony)
library(patchwork)
library(tidyr)
library(ggplot2)
library(Seurat)pbmc.stim, pbmc.ctrl是2个批次的单细胞数据。
data("pbmc_stim")
pbmc <- CreateSeuratObject(counts = cbind(pbmc.stim, pbmc.ctrl),
project = "PBMC", min.cells = 5)(2) 添加批次信息
## Separate conditions
pbmc@meta.data$batch <- c(rep("STIM", ncol(pbmc.stim)), rep("CTRL", ncol(pbmc.ctrl)))(3) 数据的归一化
pbmc <- pbmc %>%
NormalizeData(verbose = FALSE)
VariableFeatures(pbmc) <- split(row.names(pbmc@meta.data), pbmc@meta.data$batch) %>% lapply(function(cells_use) {
pbmc[,cells_use] %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
VariableFeatures()
}) %>% unlist %>% unique(4) PCA降维
pbmc <- pbmc %>%
ScaleData(verbose = FALSE) %>%
RunPCA(features = VariableFeatures(pbmc), npcs = 20, verbose = FALSE)(5) 去除批次效应前的可视化
## 未进行批次效应校正
library(cowplot)
options(repr.plot.height = 5, repr.plot.width = 12)
p1 <- DimPlot(object = pbmc, reduction = "pca", pt.size = .1, group.by = "batch")
p2 <- VlnPlot(object = pbmc, features = "PC_1", group.by = "batch", pt.size = .1)
plot_grid(p1,p2)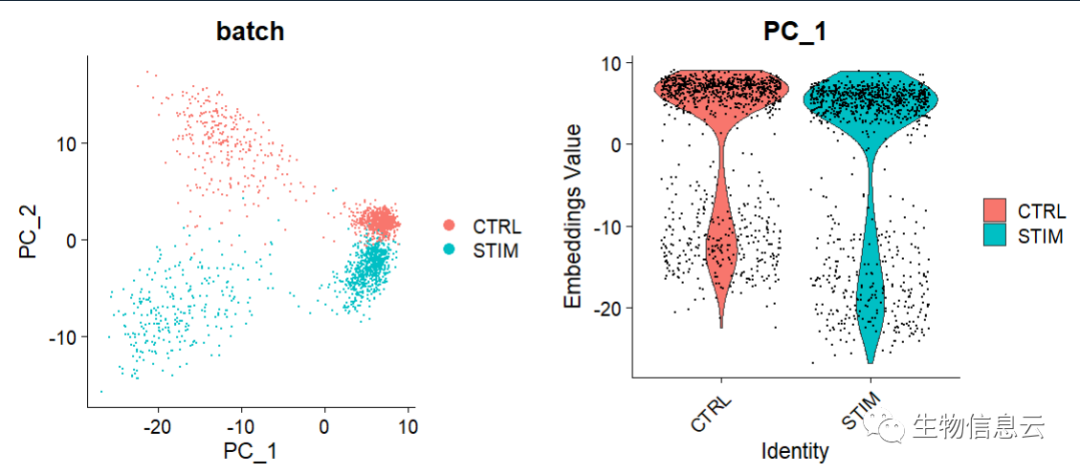
(6) 运行RunHarmony去除批次效应
bacth就是分组变量,相当于2个批次效应的单细胞数据分组信息pbmc@meta.data$bacth。
pbmc <- pbmc %>%
RunHarmony("batch", plot_convergence = TRUE,
nclust = 50, max_iter = 10, early_stop = T)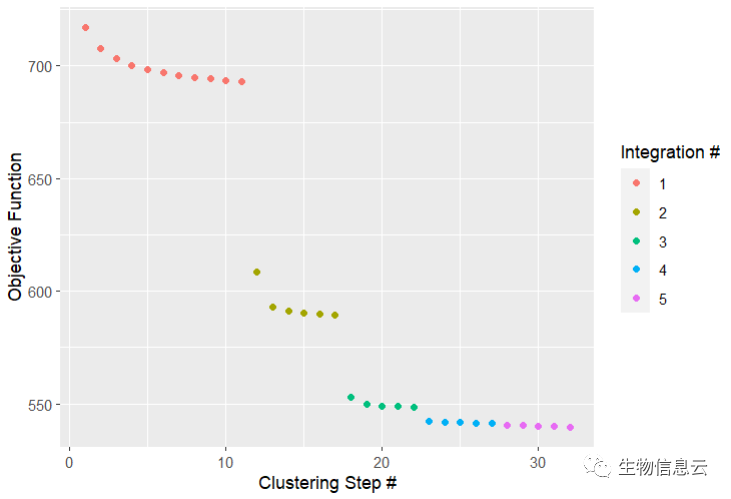
通过设置plot_converge=TRUE,Harmony将生成一个显示整合流程的目标图。每个点代表经过一轮聚类后测量的成本。不同的颜色代表由max_iter控制的不同Harmony迭代(假设early_stop=FALSE)。这里max_iter=10,预期最多有10个修正步骤。
RunHarmony函数中主要参数:
group.by.vars:
- 参数类型:字符向量(character vector)
- 描述:该参数用于指定按照哪些变量对数据进行分组,以便进行整合。
- 示例:如果数据集包含批次信息和其他变量,可以将批次信息放在一个变量中,并将该变量传递给group.by.vars参数,以便根据批次信息对数据进行整合。
max.iter.harmony:
- 参数类型:数值型(numeric)
- 描述:设置Harmony算法的最大迭代次数。默认值为10。
- 示例:如果您希望算法运行更多或更少的迭代次数,可以调整此参数的值。
lambda:
- 参数类型:数值型(numeric)
- 默认值:1
- 描述:决定了Harmony整合的力度。lambda值越小,整合力度越大;反之,lambda值越大,整合力度越小。调整范围一般在0.5-2之间。
- 示例:如果您希望增强整合力度,可以将lambda值调小;如果您希望减小整合力度,可以将lambda值调大。
theta:
- 参数类型:数值型(numeric)
- 描述:Diversity clustering penalty parameter。为group.by.vars中的每个变量指定。默认theta=2。theta=0不鼓励多样性。较大的theta值导致更多样化的集群。
- 示例:根据您的数据和研究目的,可以调整theta的值以获得不同的聚类效果。
dims.use:
- 参数类型:数值型向量(numeric vector)或字符型向量(character vector)
- 描述:指定用于Harmony的PCA维度。默认情况下,使用所有PCA维度。
- 示例:如果您只对PCA的前几个维度感兴趣,可以将dims.use参数设置为一个数值向量,例如dims.use = c(1, 2)。
sigma:
参数类型:数值型(numeric)
- 默认值:0.1
- 描述:软k均值聚类的宽度。它决定了从单个细胞到聚类中心的距离。较大的sigma值导致细胞被分配到更多的聚类中,而较小的sigma值使软k均值聚类接近于硬聚类。
- 示例:根据您的数据和聚类需求,可以调整sigma的值以获得不同的聚类效果。
这些参数共同决定了Harmony算法在运行时的行为和结果。根据您的具体需求和数据特征,您可能需要调整这些参数以获得最佳的分析效果。在使用Harmony包时,请确保仔细阅读相关文档和教程。
RunHarmony的运行结果在pbmc@reductions$harmony中。
使用Embeddings命令访问新的Harmony embeddings。
harmony_embeddings <- Embeddings(pbmc, 'harmony')
harmony_embeddings[1:4, 1:4]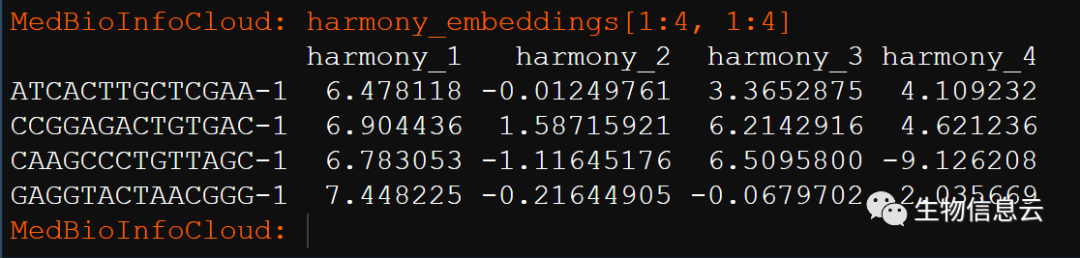
(7) 去除批次效应后的可视化
options(repr.plot.height = 5, repr.plot.width = 12)
p1 <- DimPlot(object = pbmc, reduction = "harmony", pt.size = .1,
group.by = "batch")
p2 <- VlnPlot(object = pbmc, features = "harmony_1",
group.by = "batch", pt.size = .1)
plot_grid(p1,p2)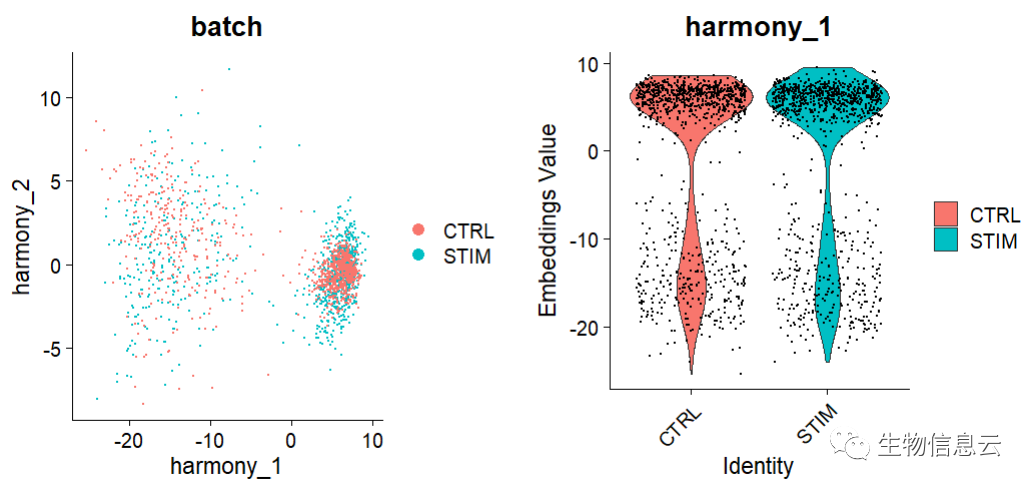
注意:Seurat对象是基于Seurat V4,现在Seurat以及V5了,主要用于解决空间多组学和转录组分析、超大规模数据量(上百万细胞,甚至更多)分析,以及函数高度集成和简化,数据结构进一步优化等。
1.3 参考资料
https://github.com/immunogenomics/harmony
https://htmlpreview.github.io/?https://github.com/immunogenomics/harmony/blob/master/doc/Seurat.html
https://htmlpreview.github.io/?https://github.com/immunogenomics/harmony/blob/master/doc/detailedWalkthrough.html
经 典 栏 目
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-12-23,如有侵权请联系 cloudcommunity@tencent.com 删除
本文分享自 MedBioInfoCloud 微信公众号,前往查看
如有侵权,请联系 cloudcommunity@tencent.com 删除。
本文参与 腾讯云自媒体同步曝光计划 ,欢迎热爱写作的你一起参与!
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号