转录组测序分析专题——质控
原创

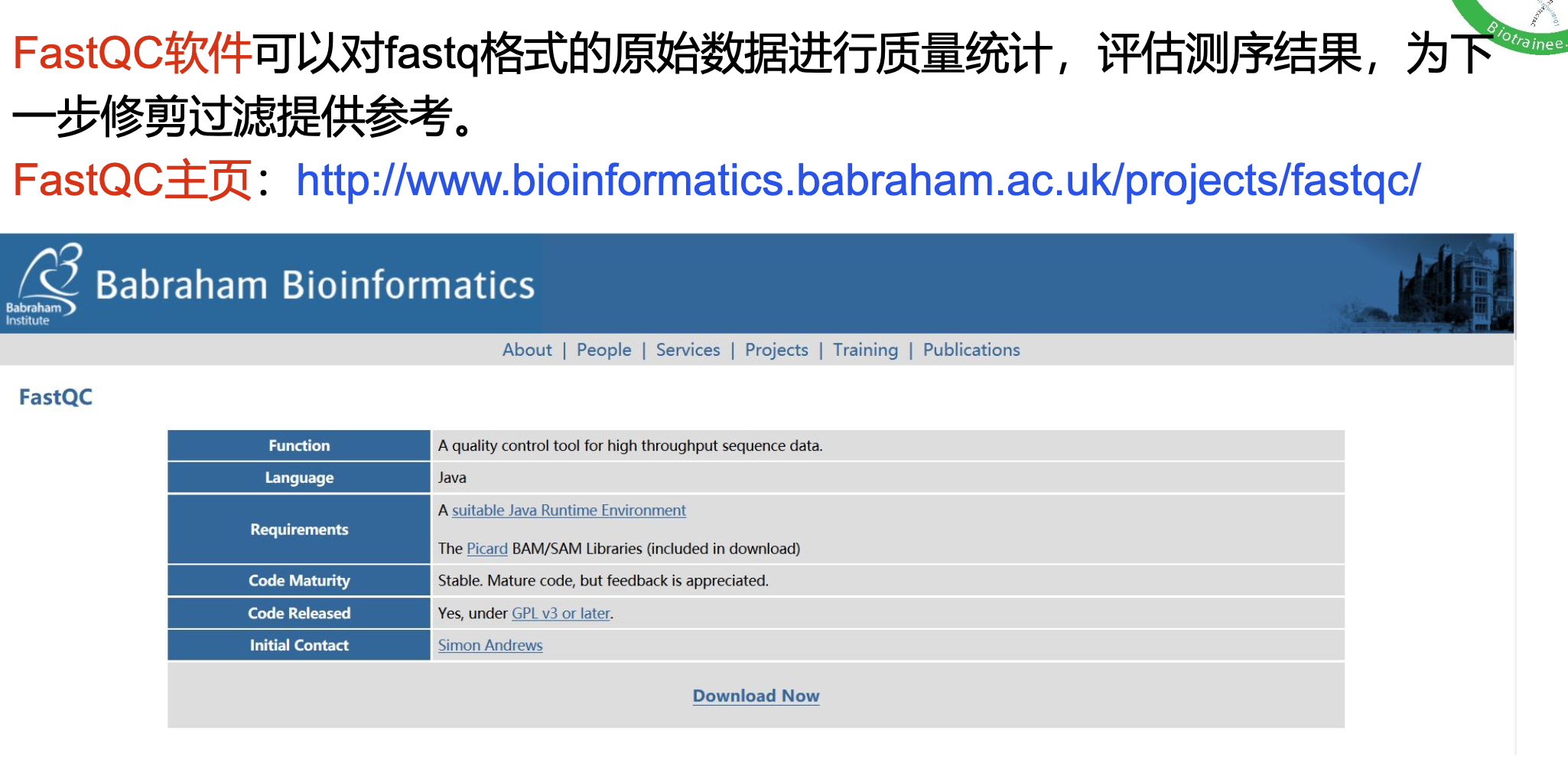
一、质控 fastqc
数据质量评估 —— fastqc

不是多有参数都有长参数和短参数两种形式;大小写敏感
目标:使用fastqc对原始数据进行质量评估
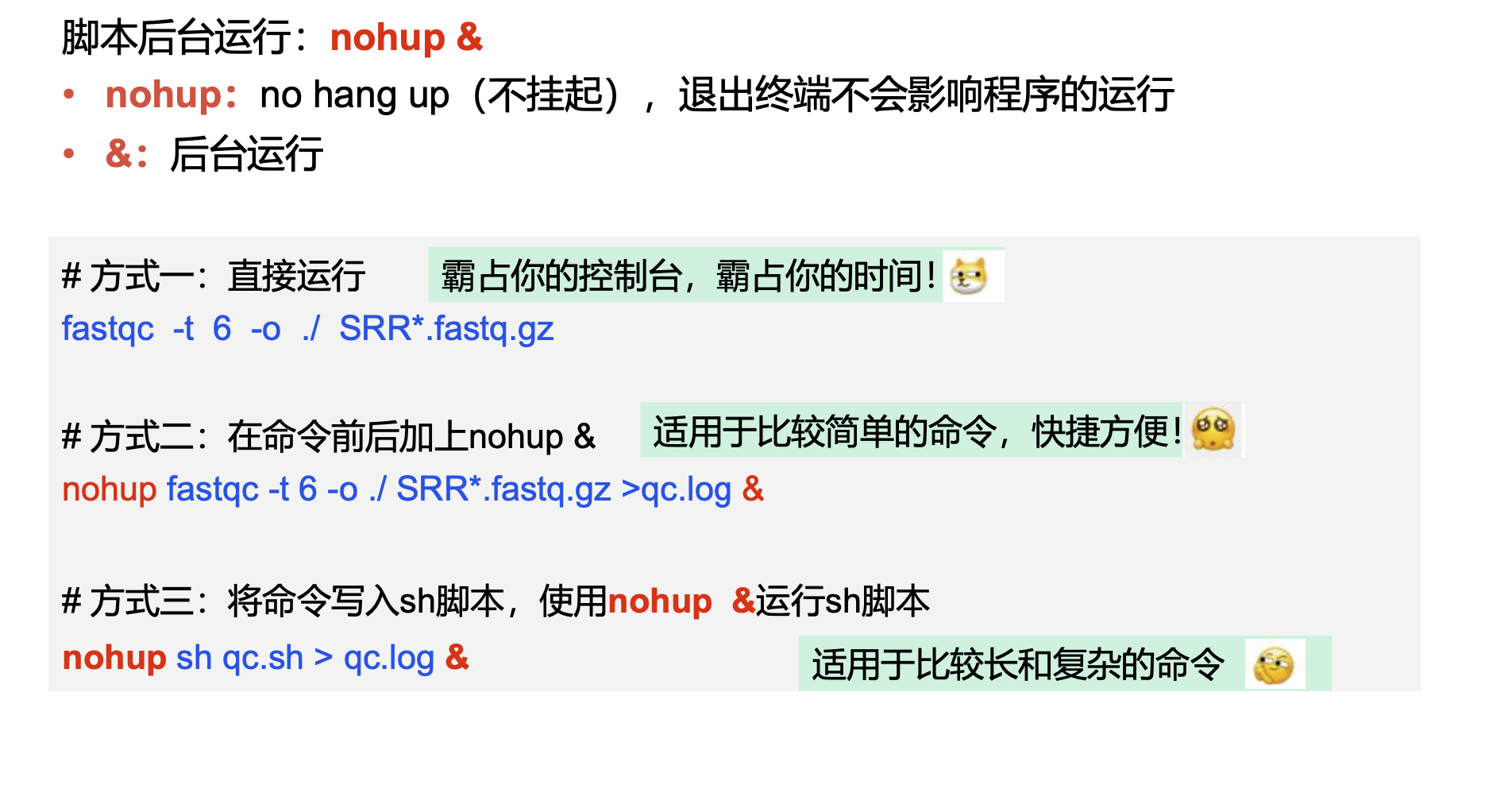
用vim将命令写入脚本qc.sh,运行脚本
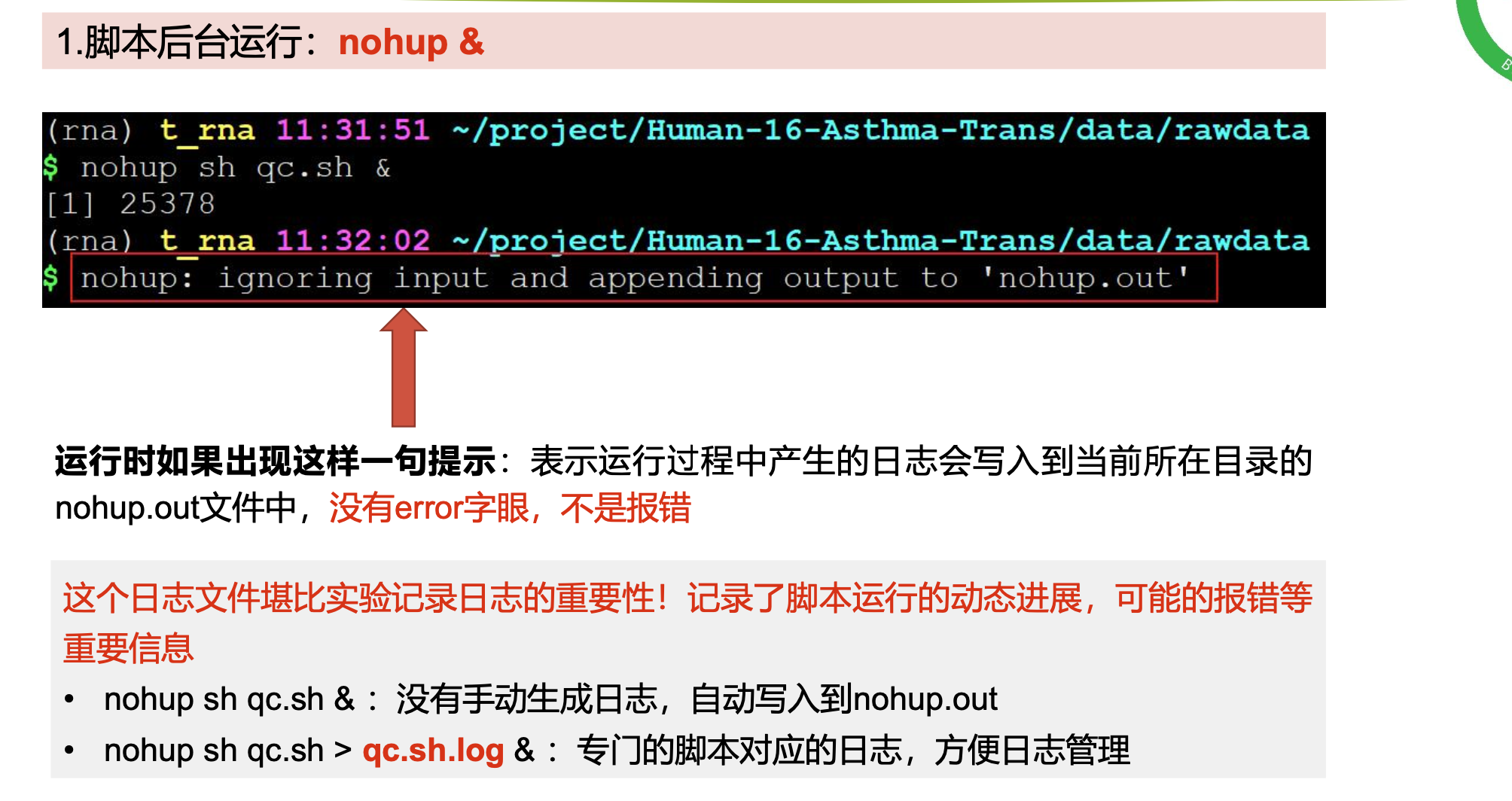
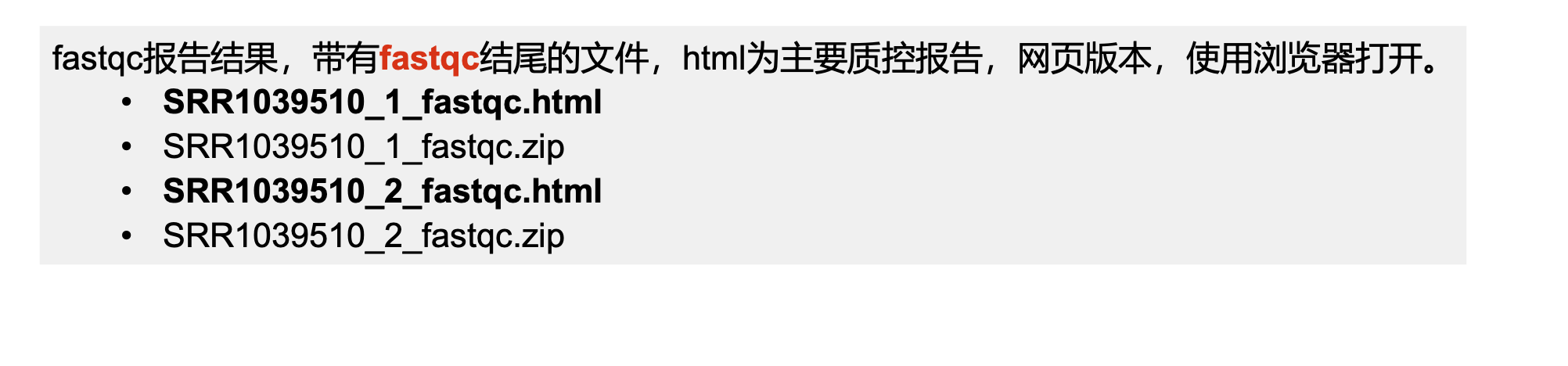
# 激活conda环境
conda activate rna
# 连接数据到自己的文件夹
# 如果上面做习题的时候已经链接过来,无需再次链接
cd $HOME/project/Human-16-Asthma-Trans/data/rawdata
ln -s /home/t_rna/data/airway/fastq_raw25000/*gz ./
# 使用FastQC软件对单个fastq文件进行质量评估,结果输出到qc/文件夹下
nohup fastqc -t 6 -o ./ SRR*.fastq.gz >qc.log & #挂到后台运行
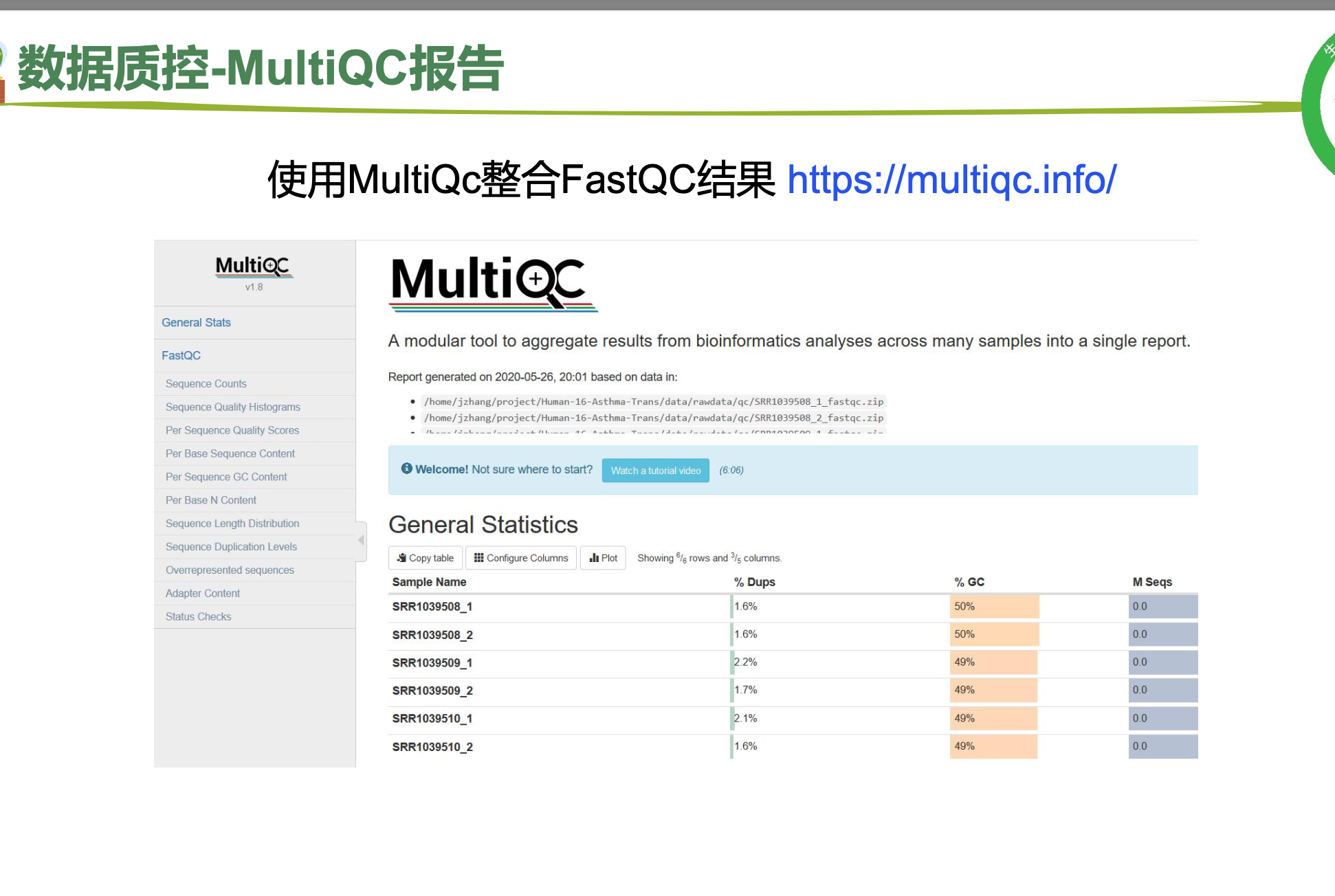
# 使用MultiQc整合FastQC结果
# 使用绝对路径运行
multiqc=/home/t_rna/miniconda3/envs/rna/bin/multiqc
fastqc=/home/t_rna/miniconda3/envs/rna/bin/fastqc
fq_dir=$HOME/project/Human-16-Asthma-Trans/data/rawdata
outdir=$HOME/project/Human-16-Asthma-Trans/data/rawdata
# 使用绝对路径运行
$fastqc -t 6 -o $outdir ${fq_dir}/SRR*.fastq.gz >${fq_dir}/qc.log
# 报告整合
$multiqc $outdir/*.zip -o $outdir/ >${fq_dir}/multiqc.log数据质控的基本数据分析
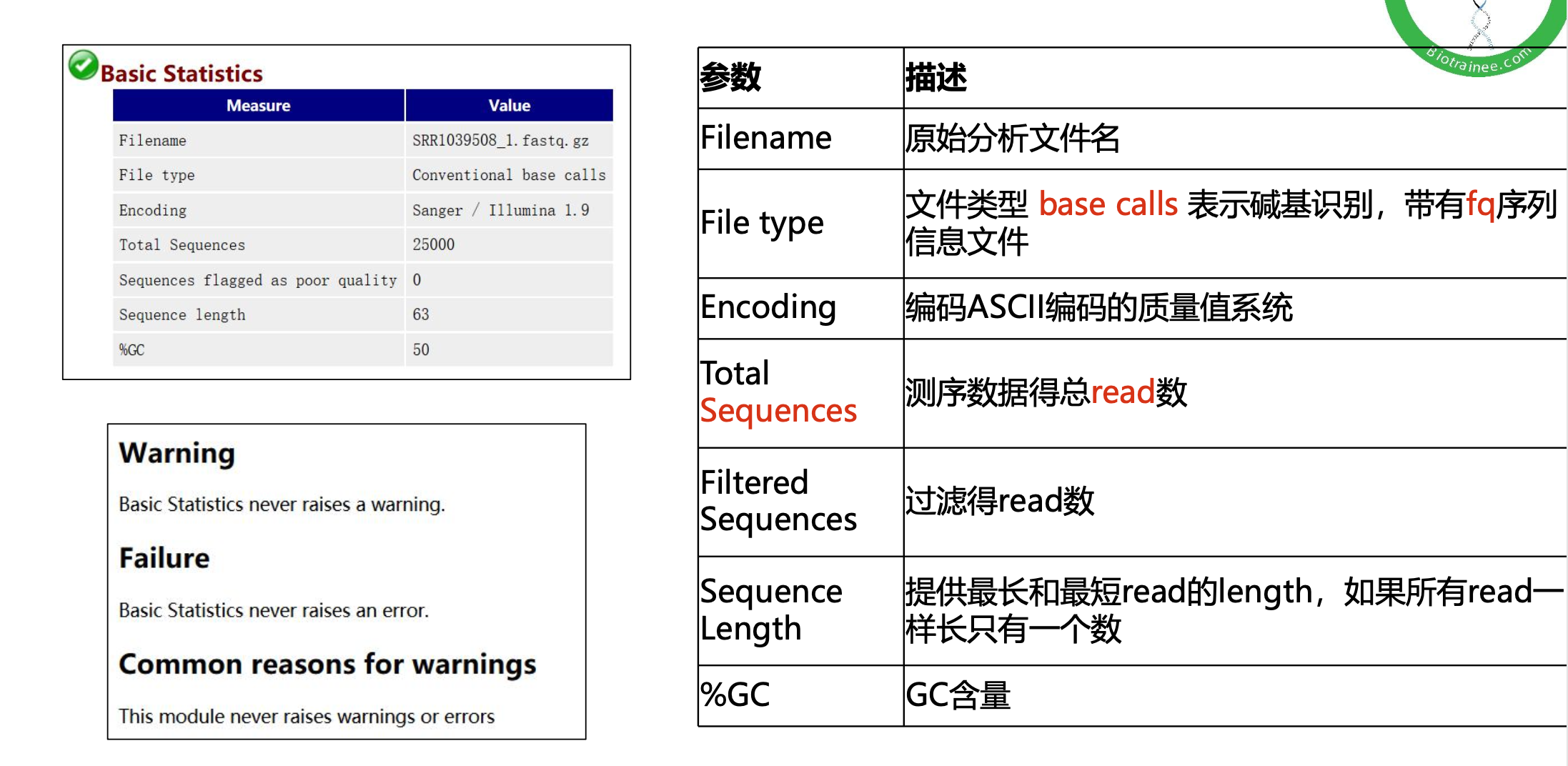
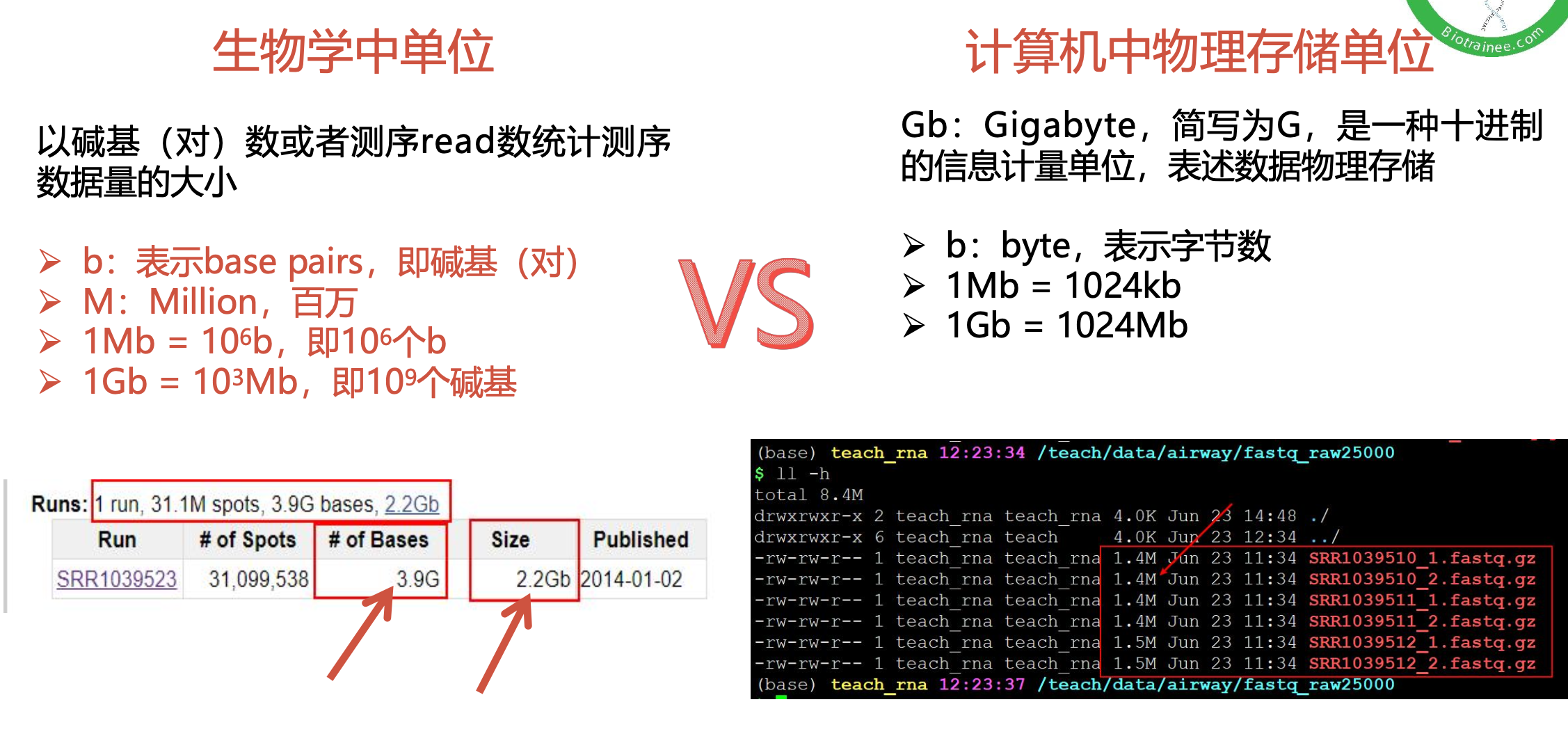
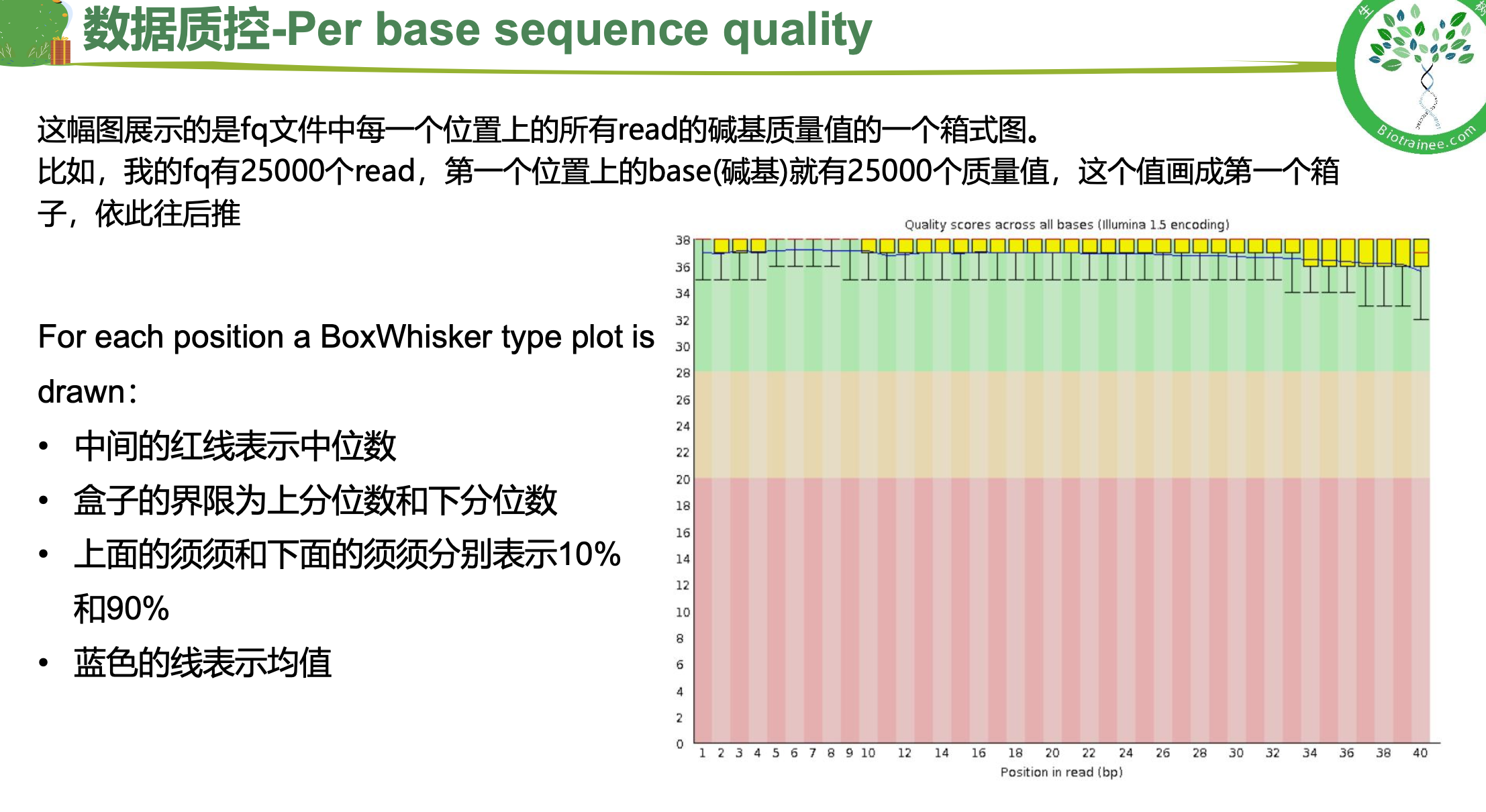
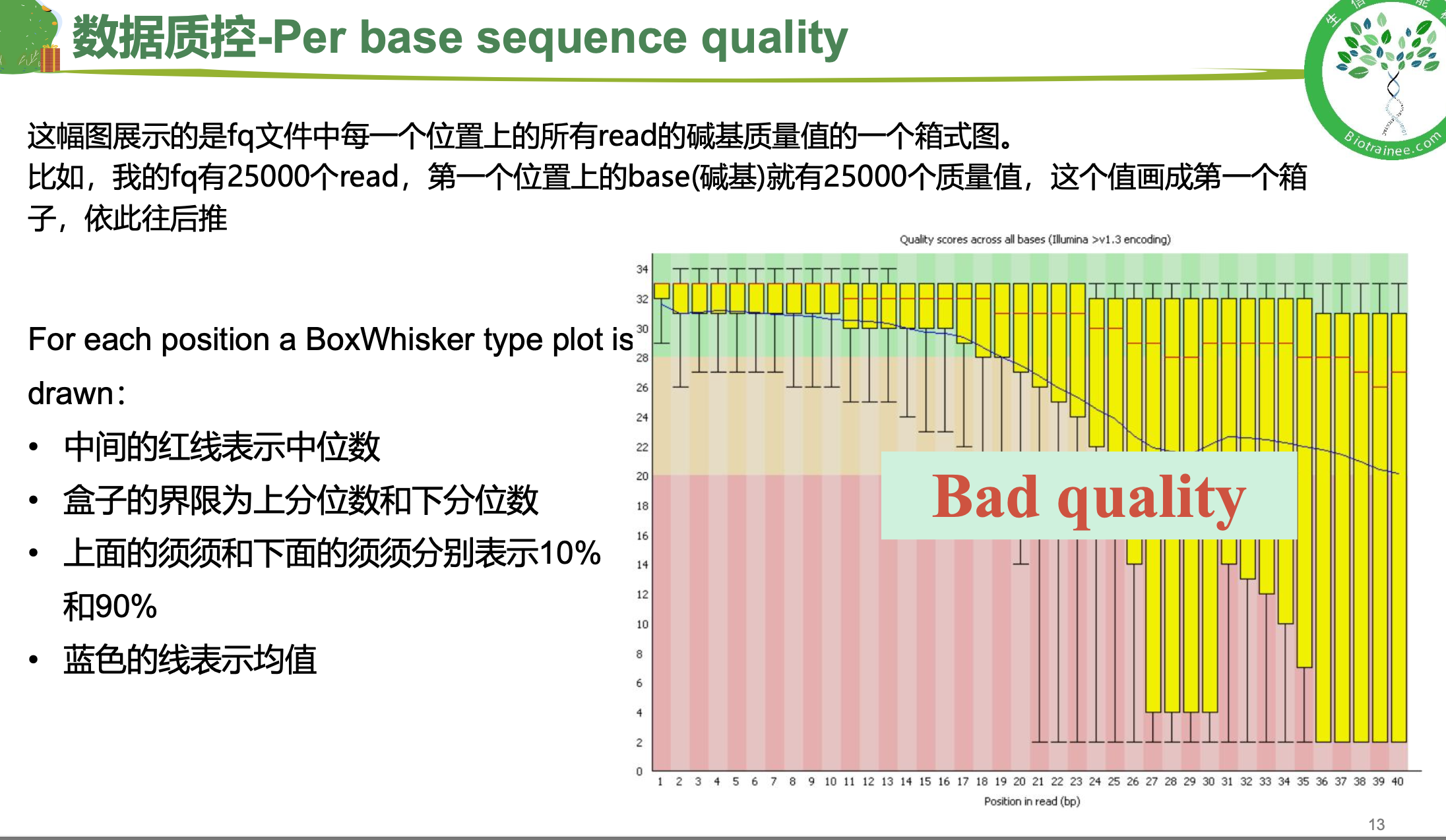
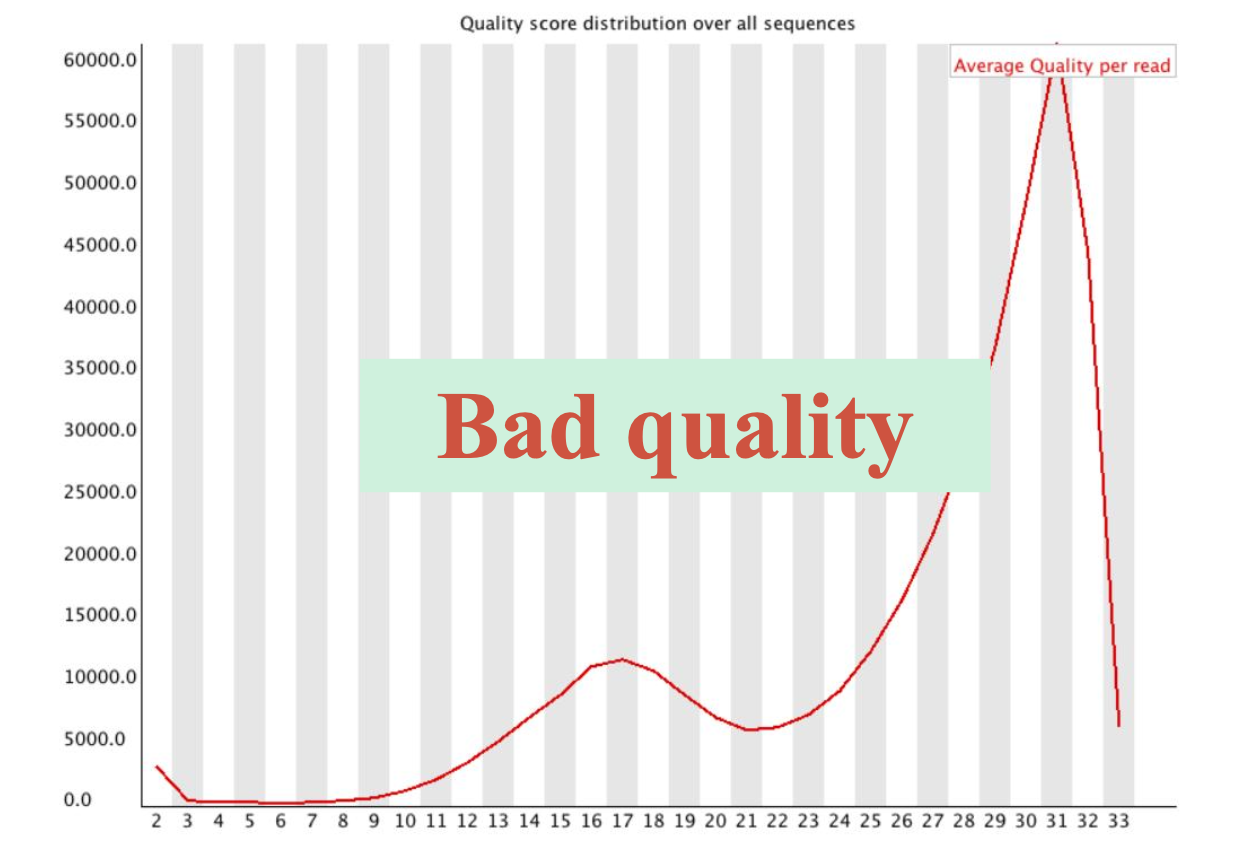
随着测序读长变长,碱基质量值下降
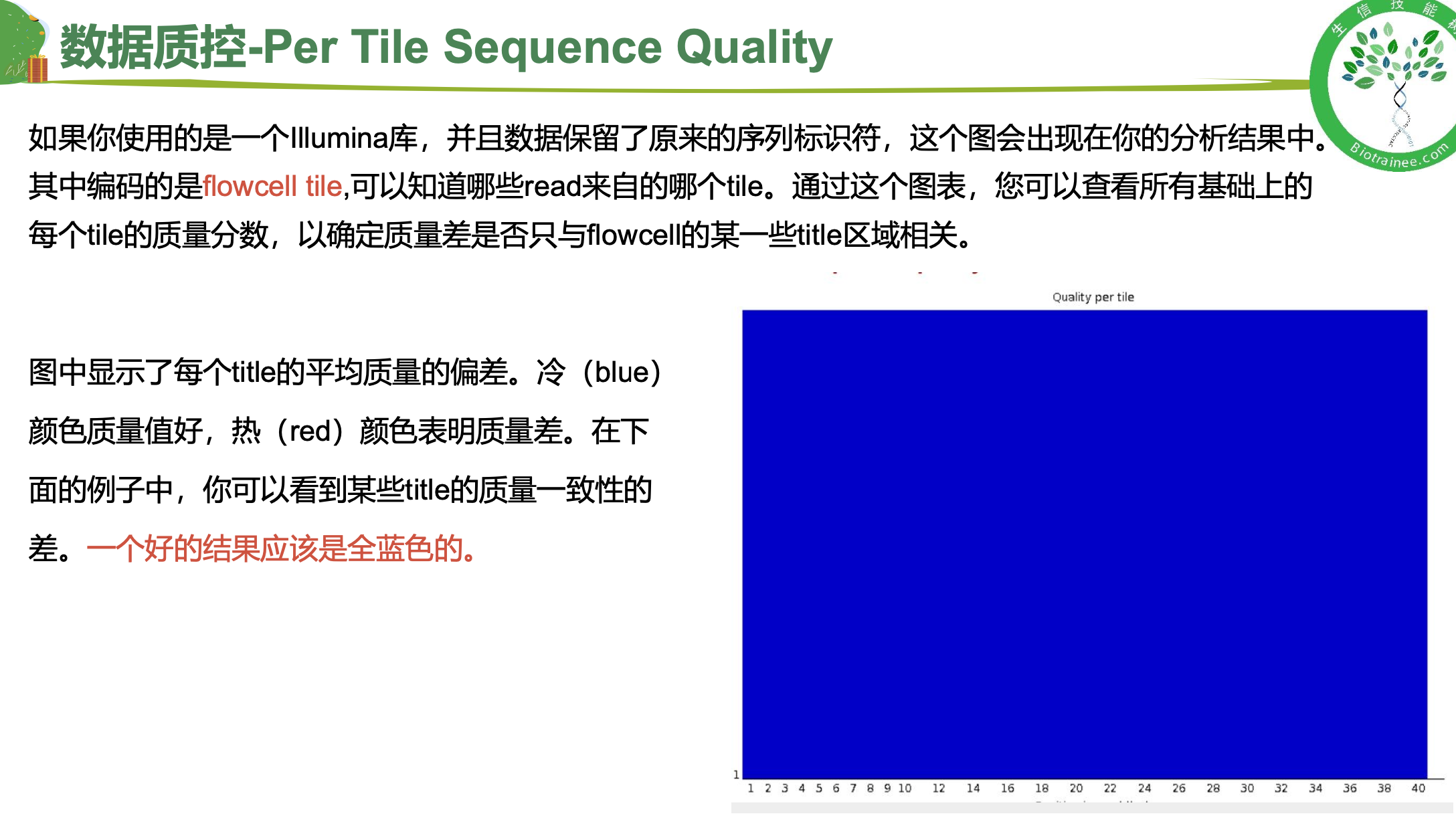
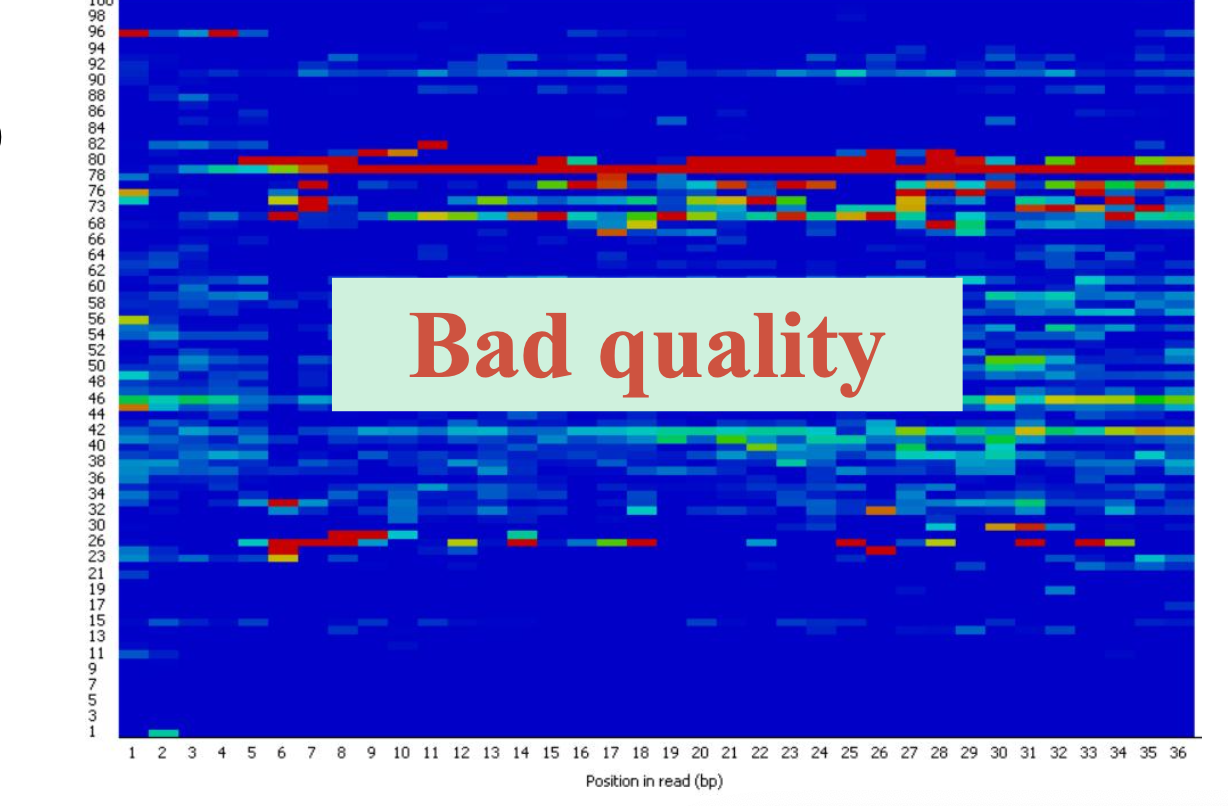
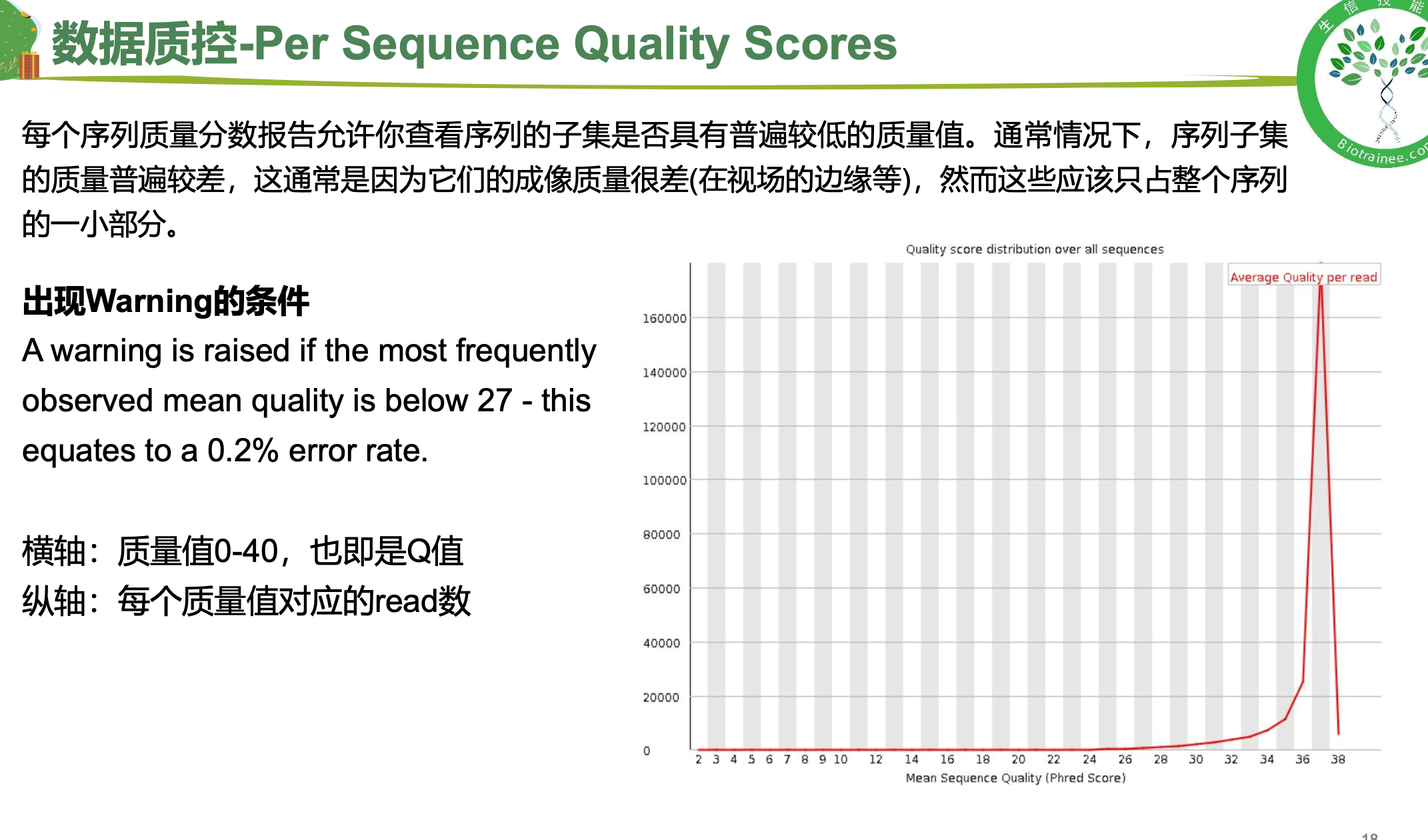
25000条序列平均碱基质量值的分布图
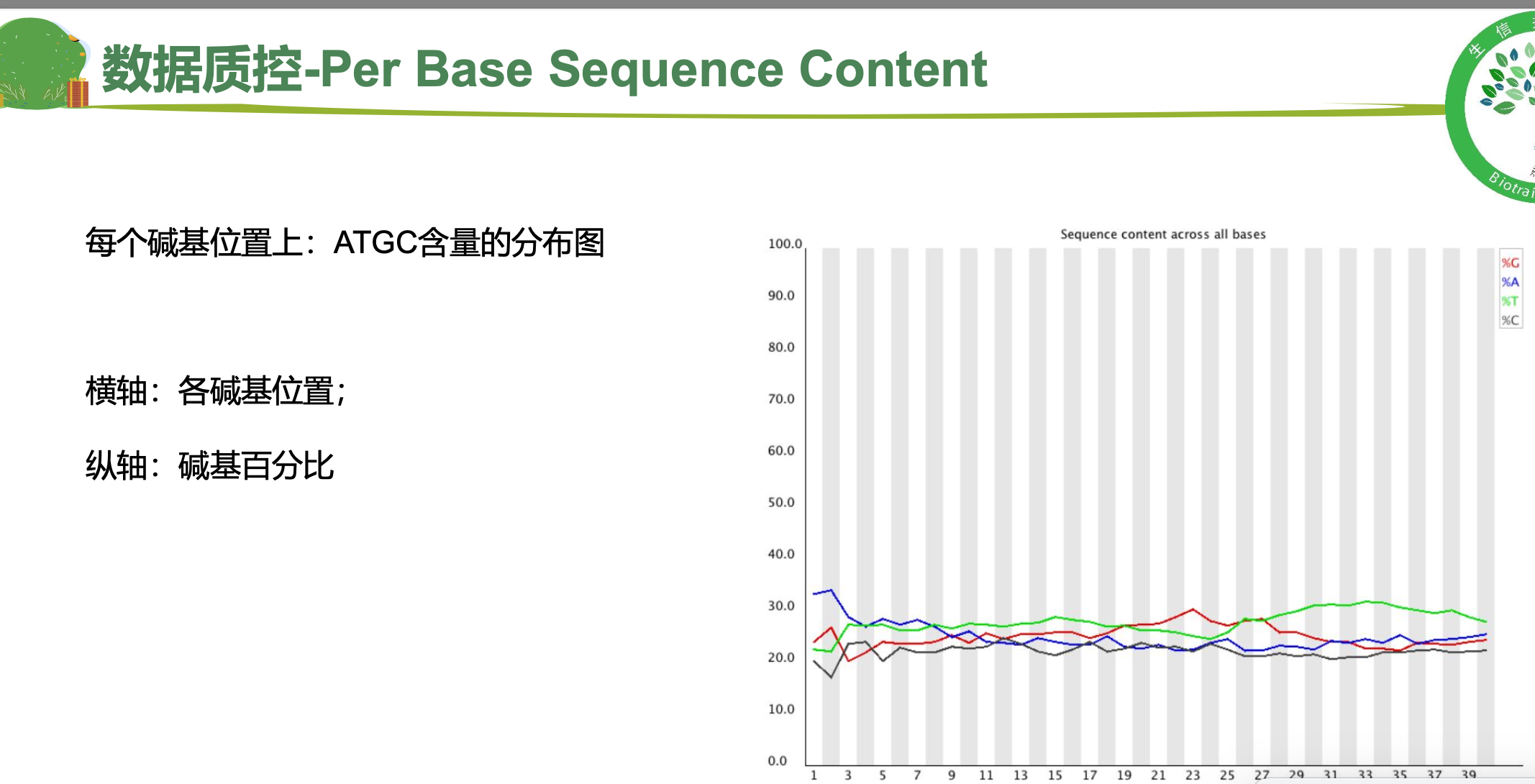
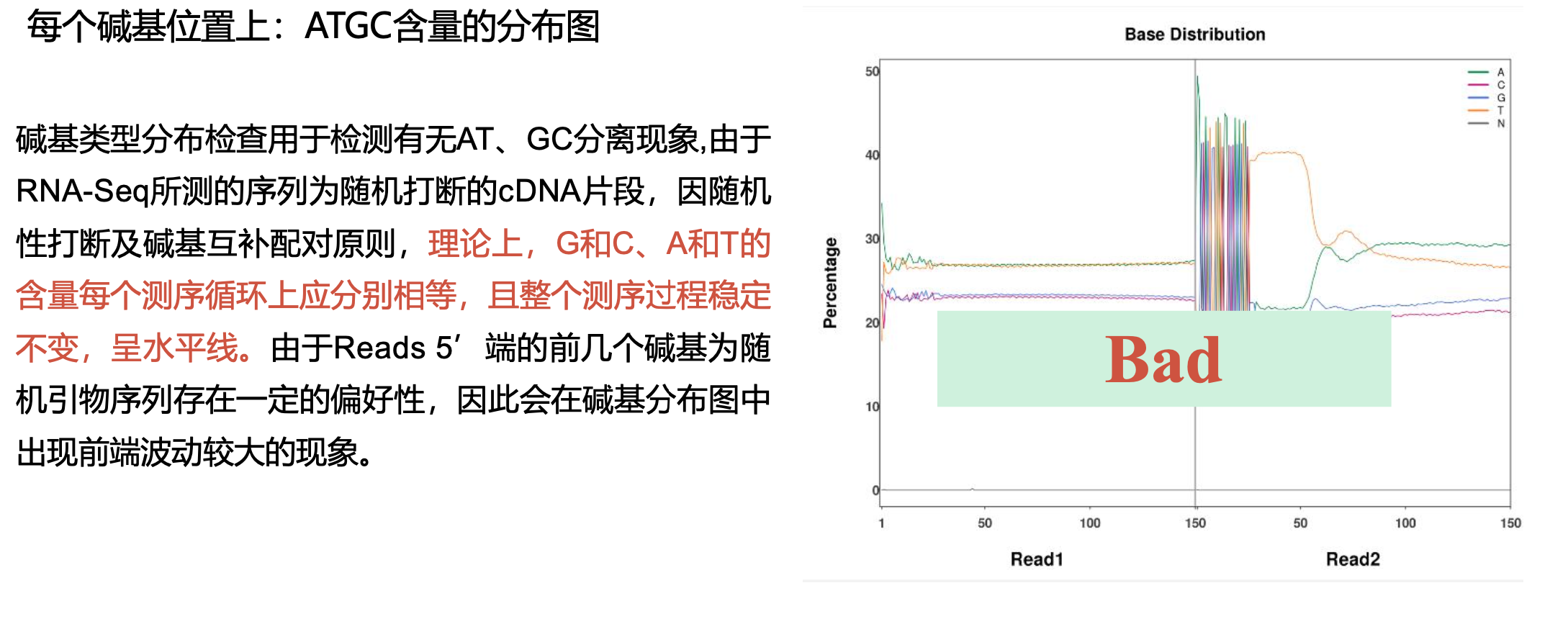
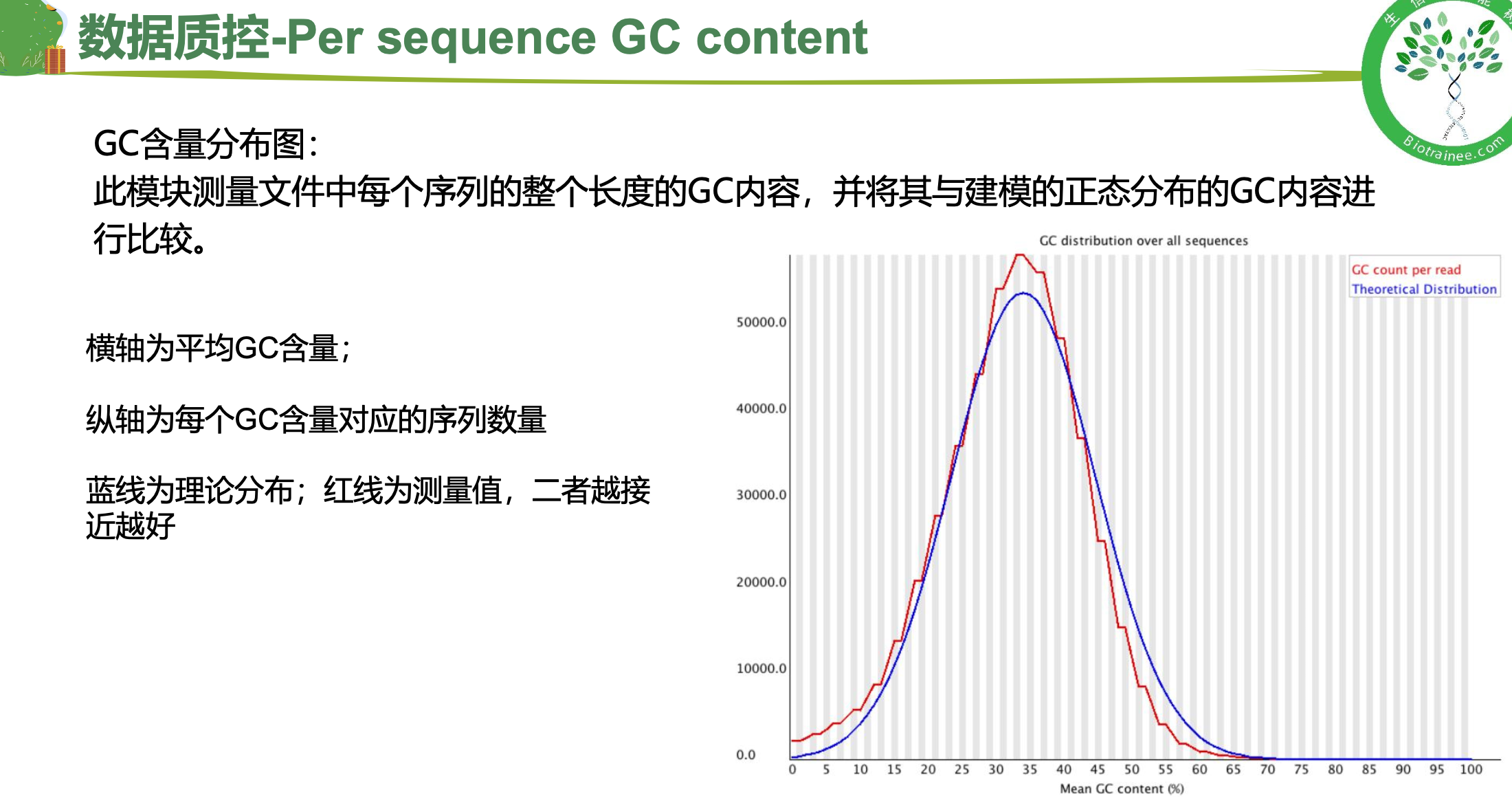
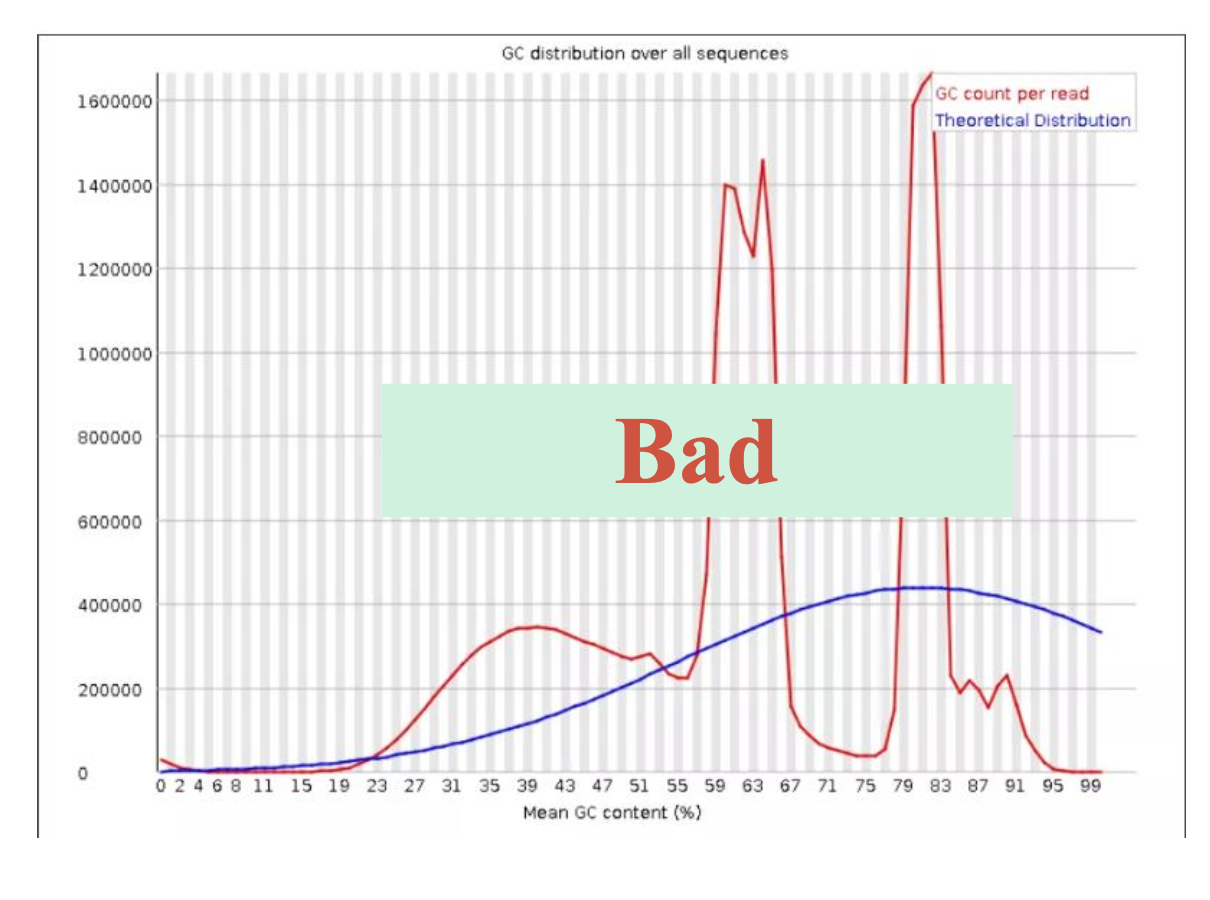
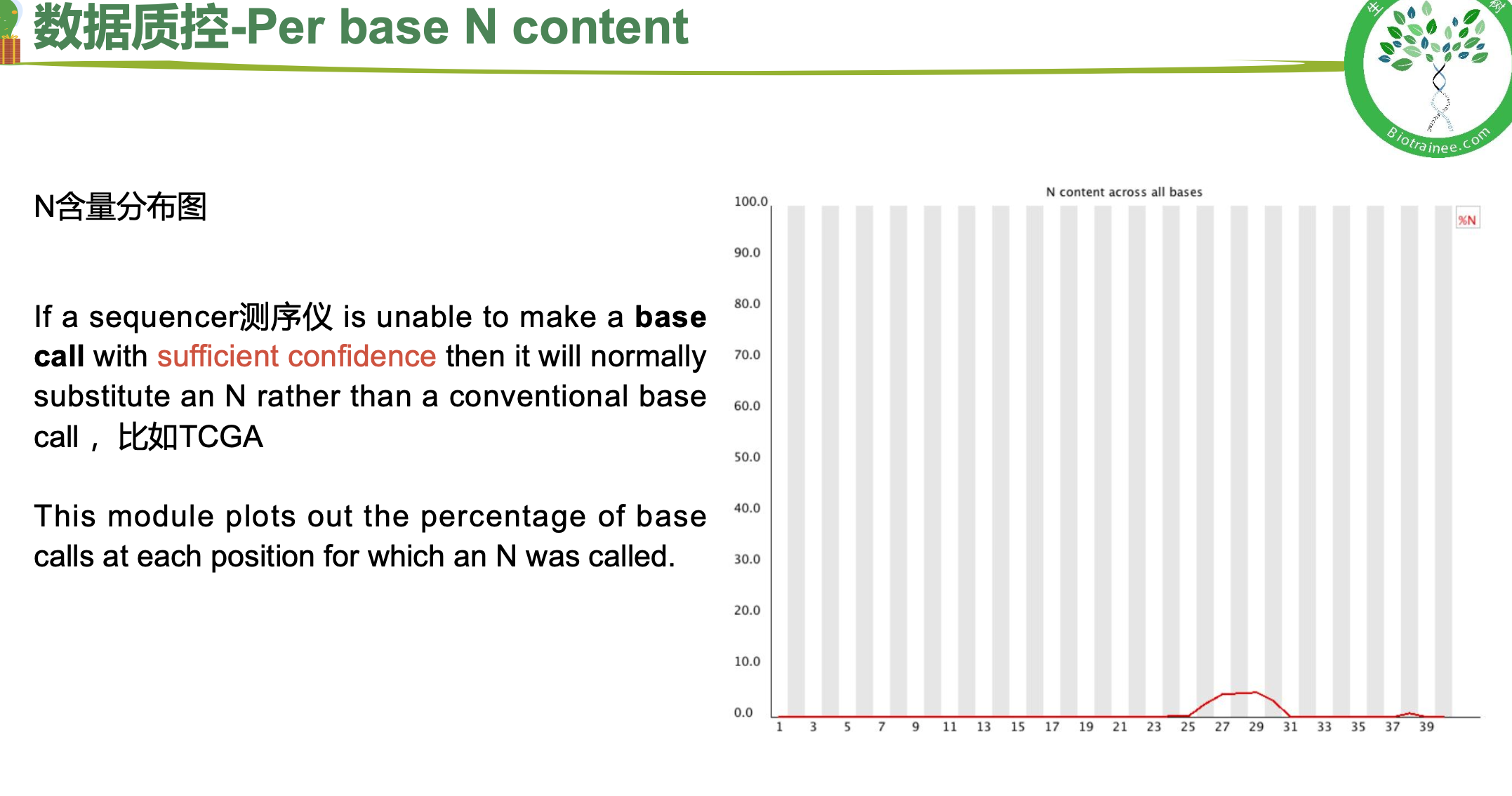
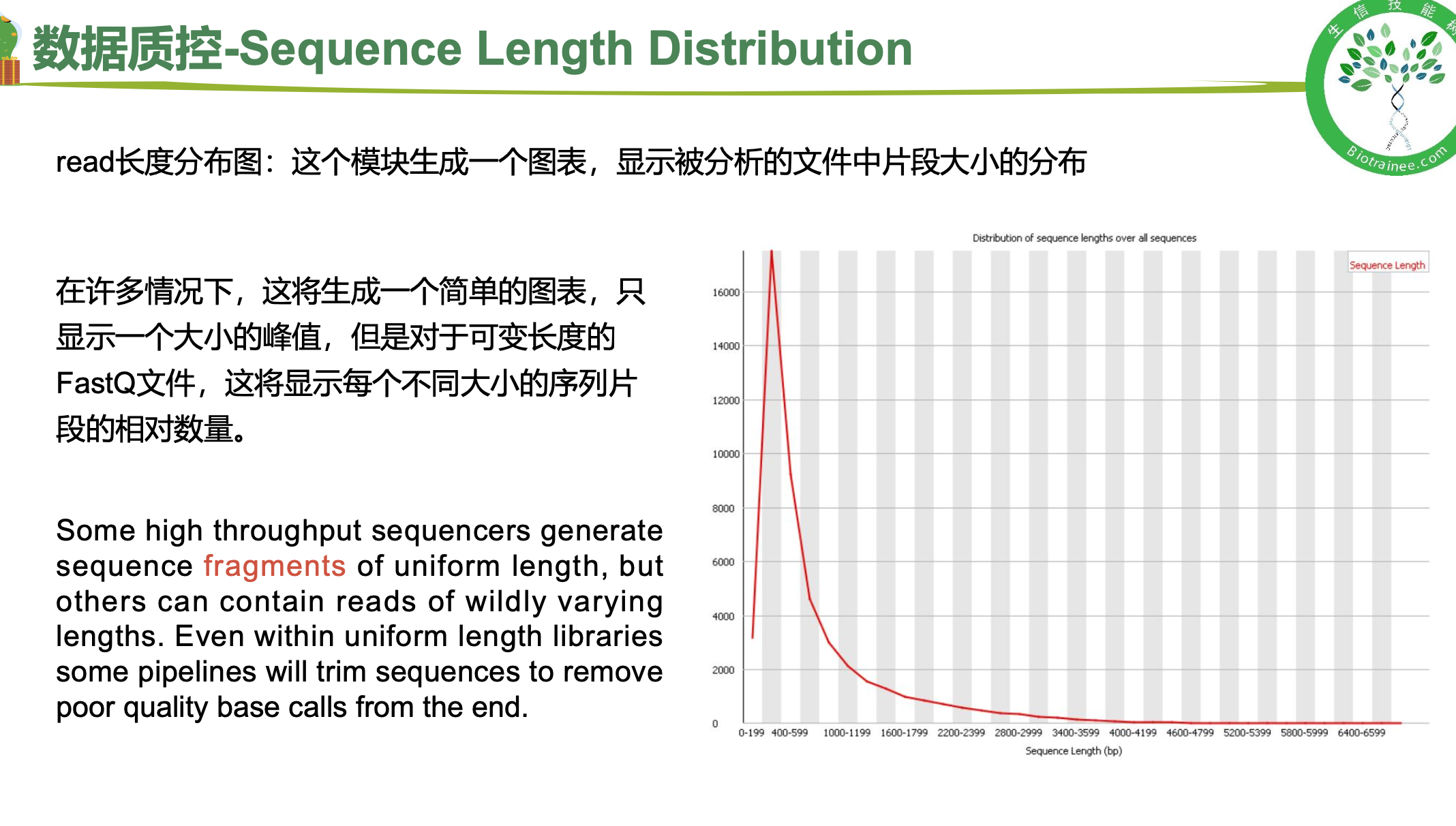
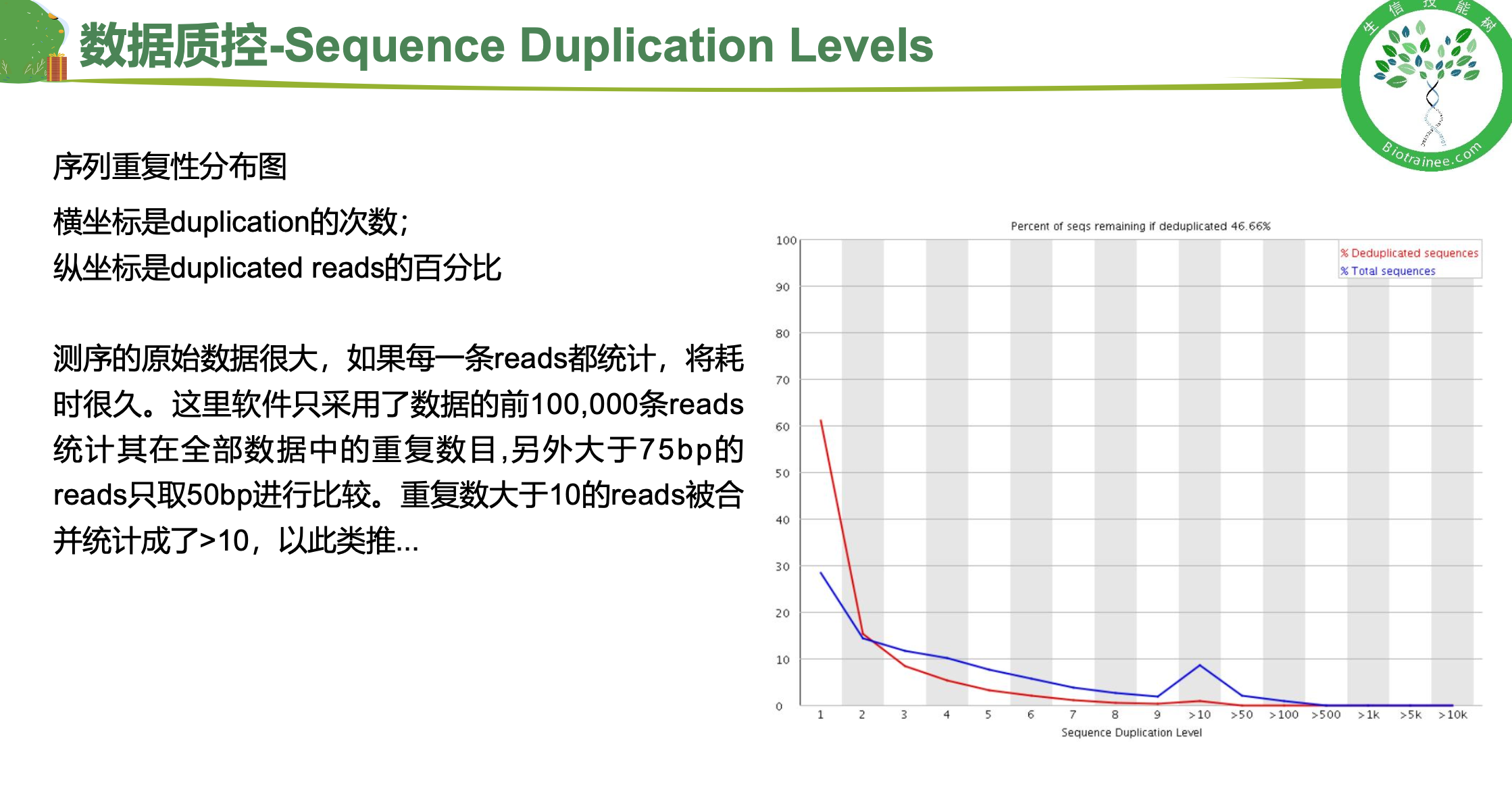

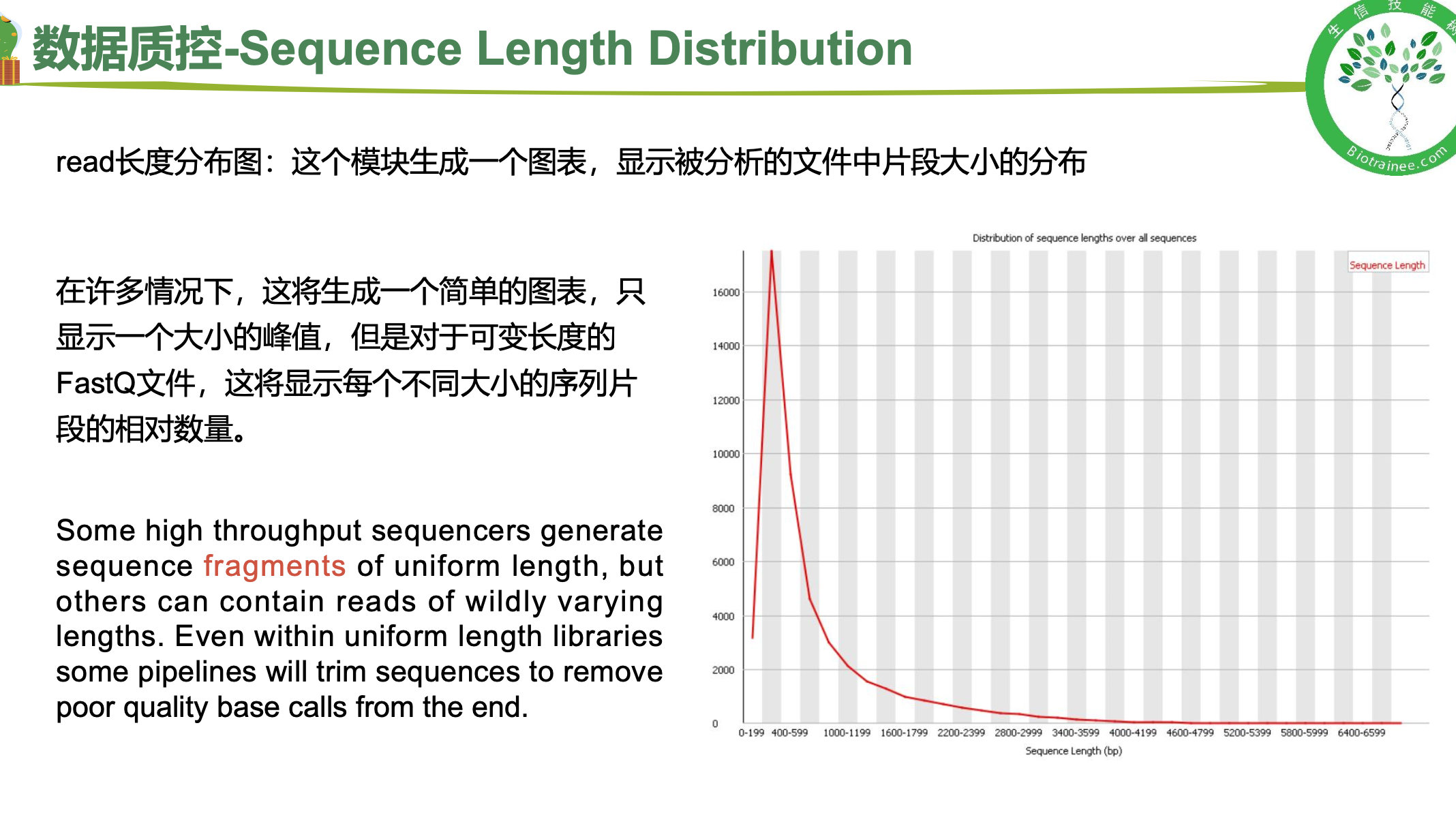
二、 trim_galore过滤低质量

tri_galore

# 激活小环境
conda activate rna
# 新建文件夹trim_galore
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
# 先生成一个变量,为样本ID
ls $HOME/project/Human-16-Asthma-Trans/data/rawdata/*_1.fastq.gz | awk -F'/' '{print $NF}' | cut -d'_' -f1 >ID
# 多个样本 vim trim_galore.sh,以下为sh的内容
rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata
cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
cat ID | while read id
do
trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${id}_1.fastq.gz ${rawdata}/${id}_2.fastq.gz
done
# 提交任务到后台 可以 用bash或者sh都行
nohup bash trim_galore.sh >trim_galore.log &
# 使用MultiQc整合FastQC结果
multiqc *.zip
## ==============================================
## 补充技巧:使用掐头去尾获得样本ID
ls $rawdata/*_1.fastq.gz | while read id
do
name=${id##*/}
name=${name%_*}
echo "trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${name}_1.fastq.gz ${rawdata}/${name}_2.fastq.gz "
done
jobs具有当前窗口时效性,只能看见当前窗口进行的任务,但ps可以看到其他窗口进行的任务

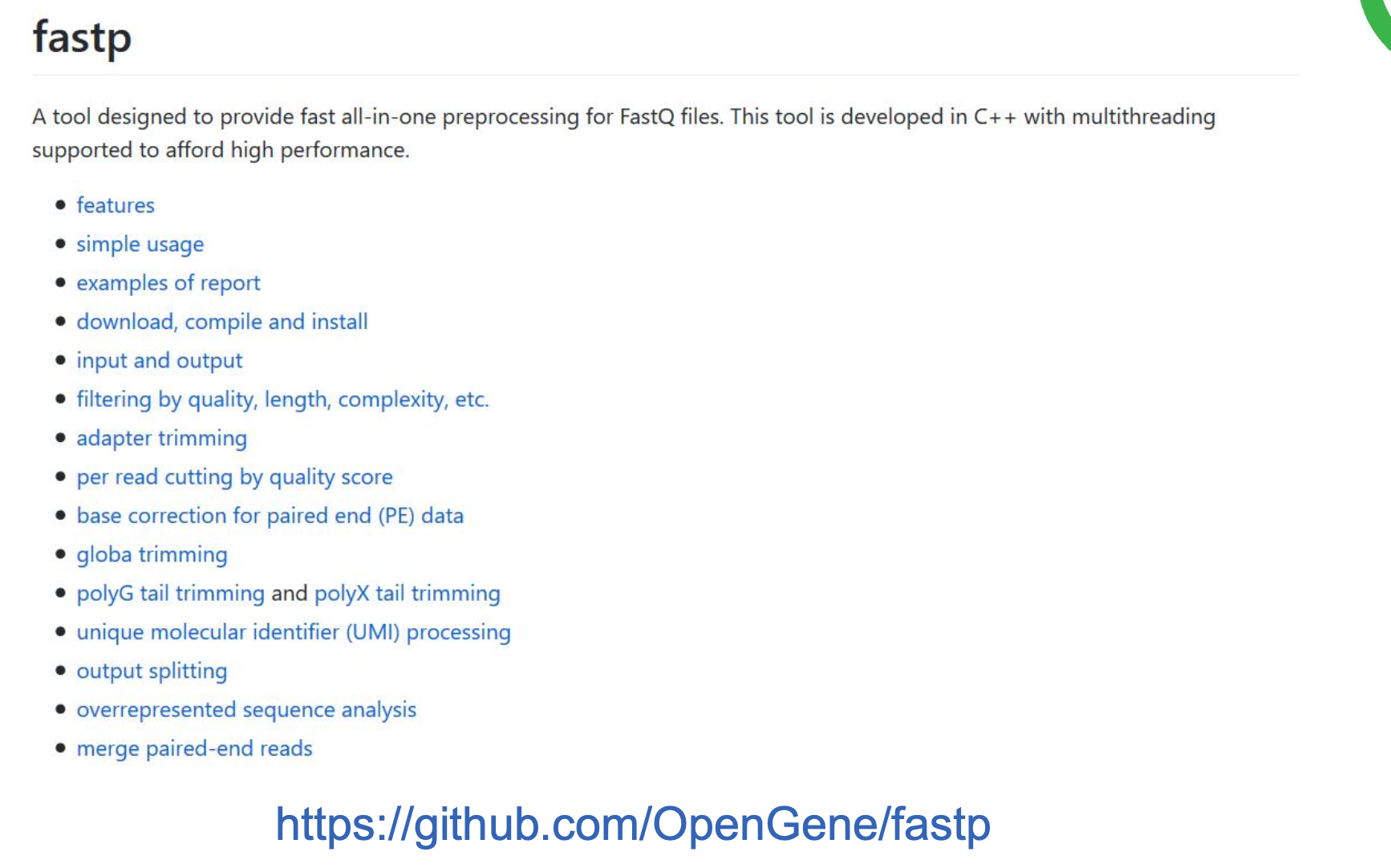
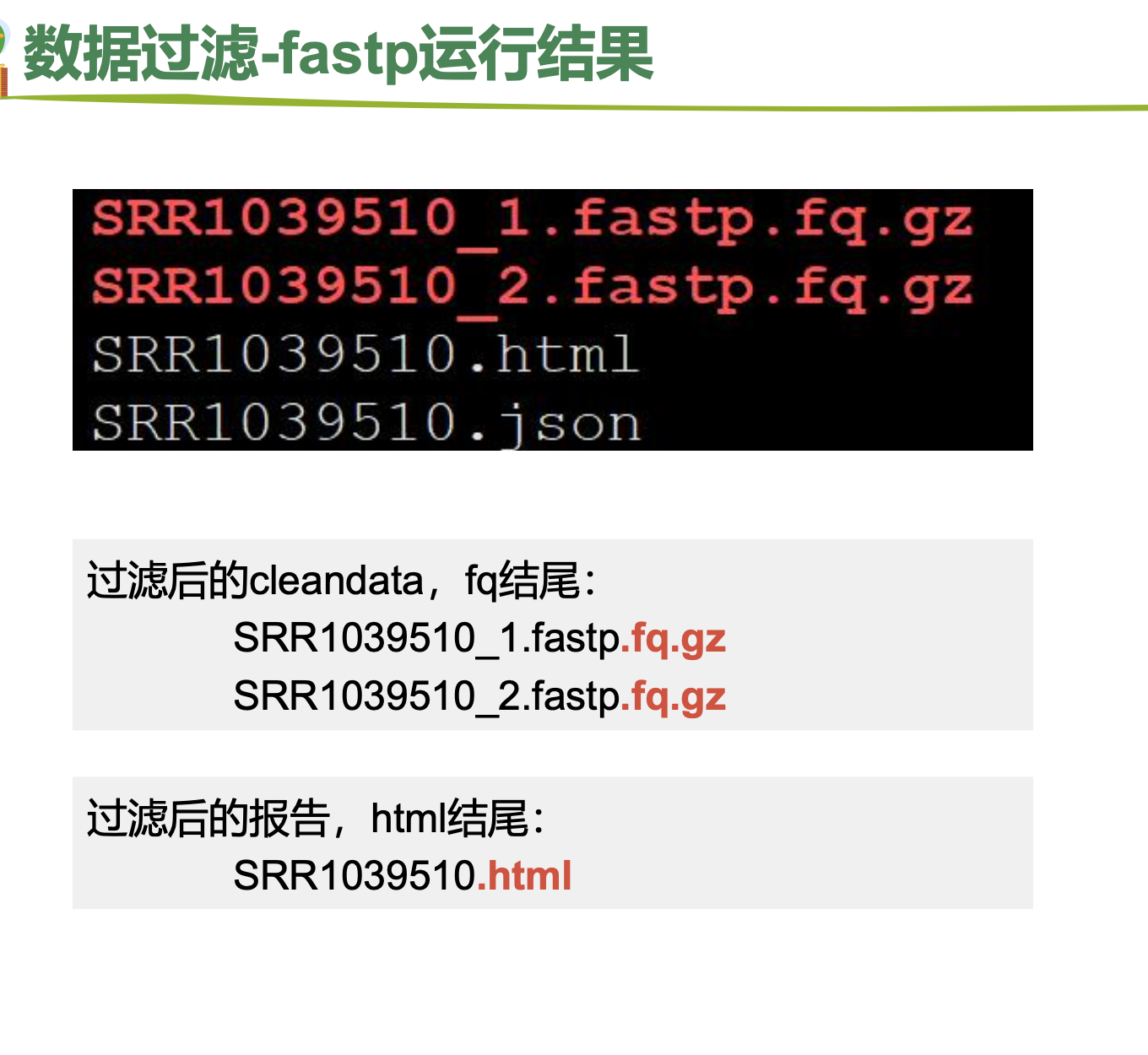
三、fastp 数据过滤
样本量很大的时候使用


\后面有空格会直接运行
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号