2023.4生信马拉松day7-R语言综合应用
原创
2023.4生信马拉松day7-R语言综合应用
原创
清南
修改于 2023-04-20 10:52:51
修改于 2023-04-20 10:52:51
本节课程大纲
六个专题——
1.玩转字符串★★★
2.玩转数据框★★★
3.条件和循环★★★★★
4.表达矩阵画箱线图★★★★
5.隐式循环★★★
6.两个数据框的连接★★
课前提示:
六个专题互不干扰互相独立,学会一个算一个;
发现问题的眼睛+面对困难的信心+解决问题的能力!
本节课涉及到的R包主要有三个:stringr、dplyr、tidyr
课前准备工作:
options("repos" = c(CRAN="http://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
if(!require(tidyr))install.packages("tidyr",update = F,ask = F)
if(!require(dplyr))install.packages("dplyr",update = F,ask = F)
if(!require(stringr))install.packages('stringr',update = F,ask = F)
if(!require(tibble))install.packages('tibble',update = F,ask = F)
library(tidyr)
library(dplyr)
library(stringr)
library(tibble)专题1 玩转字符串★★★
stringr包含几十个函数,本节课主要讲以下6个:

注:本文中未单独说明的所有图片均引用自 生信技能树小洁老师
1. str_length() 检测字符串长度
str_length()数的是引号里面有多少个字母;
v.s. length()数的是向量里面有多少个元素;
rm(list = ls())
if(!require(stringr))install.packages('stringr')
library(stringr)
x <- "The birch canoe slid on the smooth planks."
x
### 1.检测字符串长度
str_length(x)
length(x)2. str_split(字符串,"拆分符号") 拆分字符串

示例:按空格拆分x
-(1)拆分之后成为了了列表,列表的每个元素对应原来的每个元素拆分的结果
-(2)列表使用不方便——simplify = T简化结果,简化成矩阵
-(3)注意:之前提到过,矩阵的某一列不能单独转换数据类型,需要把矩阵转换成数据框再转换某列的数据类型;或者把这列单独提取出来再转换其数据类型;
### 2.字符串拆分
str_split(x," ")
x2 = str_split(x," ")[[1]];x2 #按空格拆分x,注意没有赋值就没有发生过!
y = c("jimmy 150","nicker 140","tony 152")
str_split(y," ")
str_split(y," ",simplify = T) #简化拆分结果,简化成返回矩阵而不是列表3. str_sub() 按位置提取字符串子集
str_sub(x,5,9) #提取x的第5到9个字符4. str_detect() 字符串检测【重要】
-(1)判断每个字符串含不含有某个字母或者多个字母的组合;
-(2)判断之后得到一个与x2相等的逻辑值向量;
-(3)可以用来做“根据逻辑值提取x的子集”;
str_detect(x2,"h") #判断x的每个字符串含不含有某个字母或者多个字母的组合;
str_starts(x2,"T") #判断是否以某个元素开头;
str_ends(x2,"e") #判断是否以某个元素结尾;5. str_replace()、str_replace_all() 字符替换
-(1)str_replace() :只替换匹配到的第一个目标
-(2)全部替换:str_replace_all()
x2
str_replace(x2,"o","A")
str_replace_all(x2,"o","A")6. str_remove()、str_remove_all() 字符删除
-(1)str_remove() :只删除匹配到的第一个目标
-(2)全部替换:str_remove_all()
x
str_remove(x," ")
str_remove_all(x," ")专题2 玩转数据框★★★
1. arrange() 数据框排序
-(1)arrange(test, Sepal.Length)默认按照某列对整行进行排序,不改变列与列之间的对应关系;
-(2)默认从小到大排序;要改为从大到小排序的话改成arrange(test, desc(Sepal.Length))
test <- iris[c(1:2,51:52,101:102),]
rownames(test) =NULL # 去掉行名,NULL是“什么都没有”
test
# arrange,数据框按照某一列排序
library(dplyr)
arrange(test, Sepal.Length) #从小到大
arrange(test, desc(Sepal.Length)) #从大到小2. distinct()去重复
# distinct,数据框按照某一列去重复
distinct(test,Species,.keep_all = T)其中.keep_all = T表示“按'Species'列去完重复后保留所有列”,其默认设置是.keep_all = F,表示“按'Species'列去完重复后只保留'Species'列”;
3. mutated()数据框新增一列
mutate(test, new = Sepal.Length * Sepal.Width)
#问题:新增列之后,test这个数据框是5列还是6列(有没有发生改变)?——没有,因为没有赋值就没有发生过!!!v.s. 以上操作根据此前学过的知识新增列的话这么写:

4.简单了解:select() 、filter()筛选列、行
5.补充知识:管道符%>%
-(1)当遇到连续的步骤时:多次赋值,会产生多个中间的变量;
-(2)用多次嵌套避免中间变量不直观,且容易出错;
——设置彩虹括号,可以在多层嵌套时看清楚哪个括号和哪个括号是一对:
options -- code -- display --use rainbow parentheses
-(3)用管道符%>%可以更加简洁明了;%>%表示向后传递,把管道符前面所有的东西作为后一个函数的第一个参数;管道符号永远在中间,后面一定有东西;
# 连续的步骤
# 1.多次赋值,产生多个中间的变量
x1 = select(iris,-5) #取iris除了第5列的所有列
x2 = as.matrix(x1)
x3 = head(x2,50) #取x2的前50行
pheatmap::pheatmap(x3) #用x3画热图
# 2. 嵌套,代码不易读
pheatmap::pheatmap(head(as.matrix(select(iris,-5)),50))
# 3.管道符号传递,简洁明了
iris %>%
select(-5) %>%
as.matrix() %>%
head(50) %>%
pheatmap::pheatmap()专题3 条件和循环★★★★★
1. if 条件语句:如果……就……

i = -1
if (i<0) print('up')
if (i>0) print('up')-(1)小括号里是一个逻辑值(TRUE or FALSE),不可以是多个逻辑值组成的向量;
-(2)当逻辑值为TRUE时执行大括号内的代码,如果为FALSE就不执行;
-(3)如果要执行的代码只有一行可以不加大于号;
-(4)实例:安装R包的满分操作——根据一个包是否已安装来决定要不要安装这个包;
if(!require(tidyr)) install.packages('tidyr') #根据一个包是否library成功来决定要不要安装这个包练习7-1
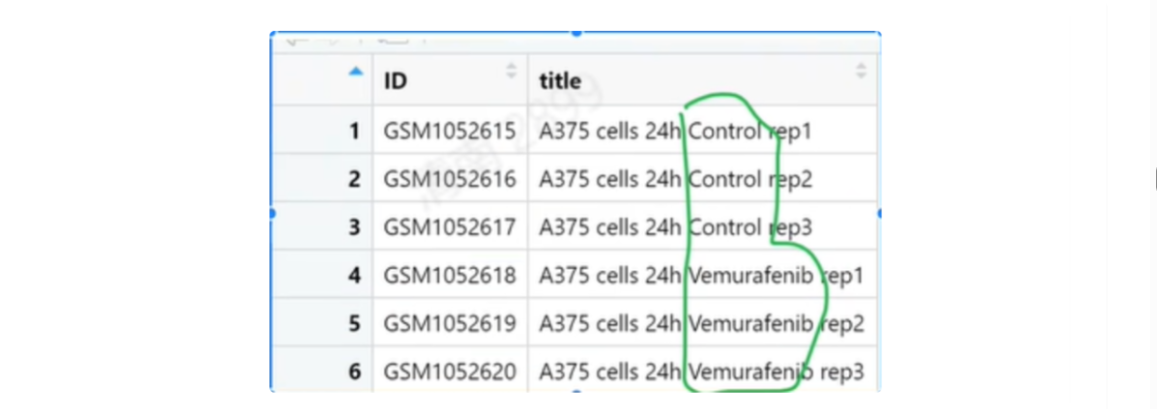
从group.csv中圈出的东西
# 1.读取group.csv,从第二列中提取圈出来的信息
g = read.csv("group.csv")
g2 = g[,2]
g2_s = str_split(g2," ",simplify = T);g2_s
g2_s[,4]
#老师的答案:
library(stringr)
a = read.csv("group.csv")
g = str_split(a$title," ",simplify = T)
g[,4]
# 2.如何把上一题结果中的Control和Vemurafenib改成全部小写?搜索一下
tolower(g2_s[,4])
str_to_lower(g[,4]) #两个函数均可
# 3.加载test1.Rdata,按照symbol列给ids数据框去重复,注意,要让ids数据真正发生修改。
load("test1.Rdata")
ids_d = distinct(ids,symbol,.keep_all = T)
dim(ids)
dim(ids_d)插播:长脚本的管理方式
-(1)用if语句控制一段代码的运行;且使用if语句,后面大括号里的代码可以折叠;
实例:用if(F){}注释掉暂时不想运行但以后还可能运行的代码(运行时把F改为T即可);直接删掉的话下次想用就得重新写;用#号大段大段注释不能折叠,影响阅读;
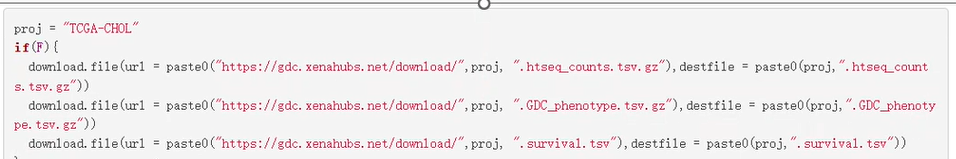
-(2)分成多个脚本,每个脚本最后保存Rdata,下一个脚本开头清空再加载,实现两个脚本之间的衔接;
#示例:
save(pd,exp,gpl,file = "step1output.Rdata")
rm(list = ls())
load("step1output.Rdata")2. if(){} else{}语句:如果……就……否则
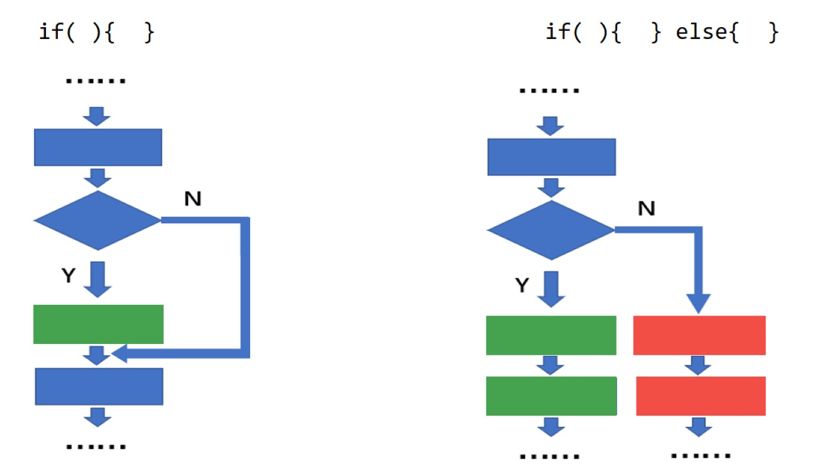
if(){} 和 if(){} else{} 的区别
i =1
if (i>0){
print('+')
} else {
print("-")
}3. ifelse()函数【重点】
-(1)三个参数:ifelse(x,yes,no)
-(2)x:逻辑值或逻辑值向量
-(3)yes:逻辑值为TRUE时的返回值
-(4)no:逻辑值为FALSE时的返回值
-(5)支持单个的逻辑值,也支持多个逻辑值组成的向量
-(6)相当于对向量的每个元素逐个进行判断,然后对判断结果F/T进行逐个替换;
i = 1
ifelse(i>0,"+","-")
x = rnorm(3)
x
ifelse(x>0,"+","-")4. ifelse()+str_detect()【王炸】
str_detect()可以检测样本中是不是含有某个字符,然后返回逻辑值,ifelse()对逻辑值T/F进行替换
samples = c("tumor1","tumor2","tumor3","normal1","normal2","normal3")
k1 = str_detect(samples,"tumor");k1
ifelse(k1,"tumor","normal")
k2 = str_detect(samples,"normal");k2
ifelse(k2,"normal","tumor")5. else if() 对多个条件进行判断
i = 0
if (i>0){
print('+')
} else if (i==0) {
print('0')
} else if (i< 0){
print('-')
}
ifelse(i>0,"+",ifelse(i<0,"-","0")) #嵌套式的ifelse:不符合大于零的条件,就再进行一步判断;练习7-2
# 1.加载deg.Rdata,根据a、b两列的值,按照以下条件生成向量x:
#a< -1 且b<0.05,则x对应的值为down;
#a>1 且b<0.05,则x对应的值为up;
#其他情况,x对应的值为no;
#统计up、down、no各出现了多少次
#我的答案:
rm(list = ls())
load("deg.Rdata")
dim(deg)
colnames(deg)
k1 = deg$a
k2 = deg$b
k3=ifelse(k1 < -1,ifelse(k2 < 0.05,"down","no"),ifelse(k1 > 1,ifelse(k2 < 0.05,"up","no"),"no"))
table(k3)
#老师的答案:
load("deg.Rdata")
k1 = deg$a< -1 & deg$b<0.05;table(k1)
k2 = deg$a>1 & deg$b<0.05;table(k2)s
x = ifelse(k1,"down",ifelse(k2,"up","no"))
table(x)6. for循环
for (i in x ) {某段代码/某种操作}
注:x 是环境中真实存在的变量,i 只是循环里的代称;
实例:
for( i in 1:4){
print(paste0("the current number is ",i))
}#批量画图
par(mfrow = c(2,2)) #把画板分成2*2的四块使其能够同时放四张图;
for(i in 1:4){
print(paste0("the current column is ",colnames(iris)[i]))
plot(iris[,i],col = iris[,5])
}#批量装包
pks = c("tidyr","dplyr","stringr")
for(g in pks){
print(paste0("package ",g," is installing"))
if(!require(g,character.only = T)) #表示a是变量名,代表包
install.packages(g,ask = F,update = F) #避免被一个需要更新的包卡住后面的代码
}专题4 表达矩阵画箱线图★★★★
1. 转换数据:把表格转换成两列数据
-(1) 第一步:转置
-(2) 第二步:把行名作为一列添加到数据中(因为ggplot2容易把行名丢掉,所以倾向于把行名作为一列)
-(3) 第三步:新增一列“group”
-(4)第四步:把宽数据变成长数据
Q:一定要先单独学会某个函数/某个包才能应用它吗?——现学就行~
# 生成一个表达矩阵
set.seed(10086) #为了让模拟分析的结果可重现,给rnorm设计一个随机数种子,保证它每次生成的随机数都是那一组;
exp = matrix(rnorm(18),ncol = 6) #新建一个矩阵,把18个随机数分成6列;
exp = round(exp,2) #取小数点后两位,不写的话默认取整
rownames(exp) = paste0("gene",1:3)
colnames(exp) = paste0("test",1:6)
exp[,1:3] = exp[,1:3]+1 #给exp的1-3列加一
exp关于set.seed():可以把它理解为给生成的随机数序列一个编号,保证其可以复现。
#处理数据
library(tidyr)
library(tibble)
library(dplyr)
dat = t(exp) %>% #转置
as.data.frame() %>% #变成数据框
rownames_to_column() %>% #把行名变成一列,
mutate(group = rep(c("control","treat"),each = 3))
#宽变长函数的最新版本:
pdat = dat%>%
pivot_longer(cols = starts_with("gene"), #把gene开头的列转换掉
names_to = "gene", #新的列名叫gene
values_to = "count") #由原来的值转换得到的那一列的列名叫count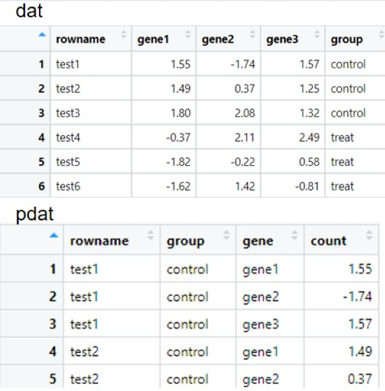
宽数据变长数据
2. 使用转换好的数据画图
#数据转换好就可以画图了
library(ggplot2)
p = ggplot(pdat,aes(gene,count))+
geom_boxplot(aes(fill = group))+
theme_bw()
p
p + facet_wrap(~gene,scales = "free") #还可以根据gene分面专题5 隐式循环★★★
1. apply()函数
-(1)apply() 处理矩阵或数据框
apply(X, MARGIN, FUN, …)
其中X是数据框/矩阵名;
MARGIN为1表示行,为2表示列,FUN是函数;
rm(list = ls())
test<- iris[1:6,1:4]
apply(test, 2, mean)
apply(test, 1, sum)
#等价于
colMeans(test)
rowSums(test)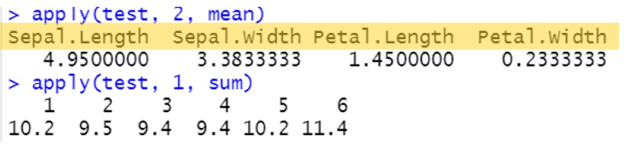
注:黄色部分是这个向量里的各个元素的名字;对行循环继承行名,对列循环继承列名;
-(2)用apply()批量画图
par(mfrow = c(2,2))
apply(iris[,1:4], 2, plot)
#如果有写不下的参数,可以继续写在apply的括号里
apply(iris[,1:4], 2, plot,col = iris[,5])
#或者也可以自定义函数
jimmy <- function(g){
plot(g,col = iris[,5])
}
par(mfrow = c(2,2))
apply(iris[,1:4], 2, jimmy)2. 如何挑出30个数里最大的五个
-(1)排序
-(2)取最后五个

3.向量/列表的隐式循环-lapply()
对列表/向量中的每个元素实施相同的操作
lapply(1:4,rnorm)
#批量画图
lapply(1:4, function(i){
plot(iris[,i],col = iris[,5])
})
#批量装包
pks = c("tidyr","dplyr","stringr")
qa = function(g){
print(paste0("package ",g," is installing"))
if(!require(g,character.only = T))
install.packages(g,ask = F,update = F)
}
lapply(pks, qa)专题6 两个数据框的连接★★
1. iner_join
两个表格按照共同的一列取交集,连接的结果是二者交集
2.left_join
以左边的表格为主,左右都有的连接保留,右边没有的用NA填充
3. right_join
以右边的表格为主
4. full_join
保留所有的,缺失的位置填充NA
5. semi_join
半连接,效果是取子集:以右边表格为参考对左边取子集
6. anti_join
保留左边表格在右边表格里没有的东西
test1 <- data.frame(name = c('jimmy','nicker','Damon','Sophie'),
blood_type = c("A","B","O","AB"))
test1
test2 <- data.frame(name = c('Damon','jimmy','nicker','tony'),
group = c("group1","group1","group2","group2"),
vision = c(4.2,4.3,4.9,4.5))
test2
library(dplyr)
inner_join(test1,test2,by="name")
right_join(test1,test2,by="name")
full_join(test1,test2,by="name")
semi_join(test1,test2,by="name")
anti_join(test1,test2,by="name")课后思考题
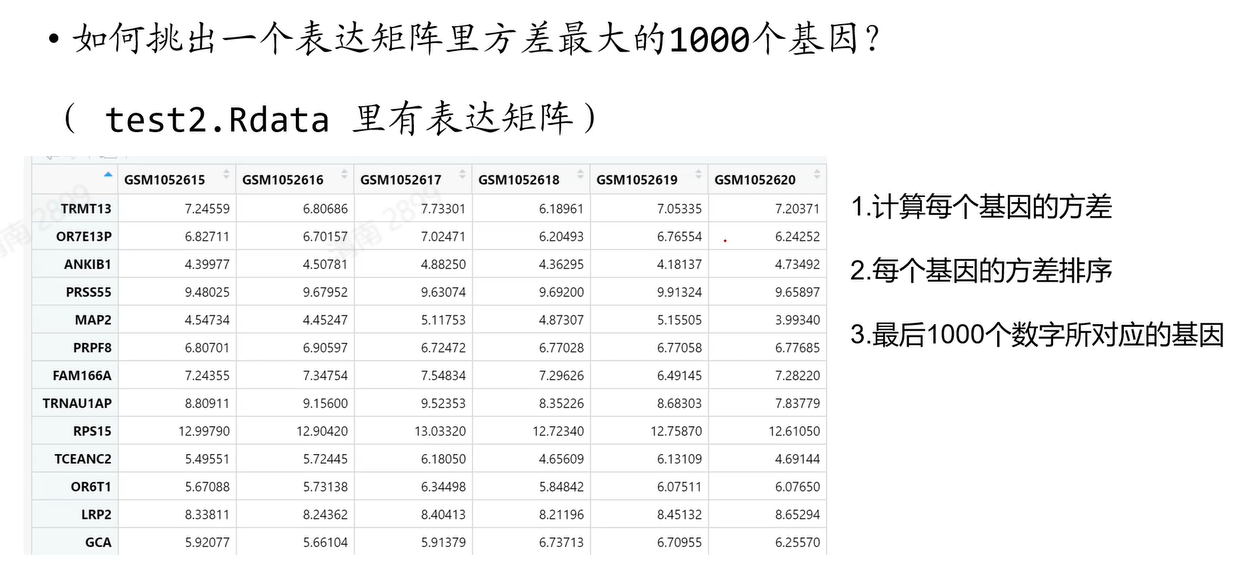
#我的答案:
load("test2.Rdata")
test = as.data.frame(test)
test$gene_var <- apply(test,1,var) #计算每行的方差并添加到test
library(tidyr)
library(tibble)
test = rownames_to_column(test,var="gene_name")
test = arrange(test,gene_var)
tail(test$gene_name,1000)
#老师的答案
load("test2.Rdata")
x=names(tail(sort(apply(test,1,var)),1000)) #看到就一行的时候我好气呀……练习7-3
#1. 加载test1.Rdata,将两个数据框按照probe_id列连接在一起,按共同列取交集
#2. 找出logFC最小的10个基因和logFC最大的10个基因(symbol列就是基因名)#我的答案:
rm(list = ls())
load("test1.Rdata")
library(dplyr)
i = inner_join(dat,ids,by="probe_id")
min10 = head(sort(i$logFC),10)
max10 = tail(sort(i$logFC),10)
min10_gene = i$symbol[i$logFC %in% min10]
max10_gene = i$symbol[i$logFC %in% max10]
min10_gene
max10_gene#老师的答案:
rm(list = ls())
load('test1.Rdata')
library(dplyr)
x = merge(dat,ids,by = "probe_id")
x2 = inner_join(dat,ids,by = "probe_id")
dim(x)
dim(x2)
x = arrange(x,logFC)
head(x$symbol,10)
tail(x$symbol,10)
head(x$logFC,10)我发现我的答案和老师的答案给出的基因名是相同的,但顺序不同;对比之后发现我的是按排序前原本的先后顺序列出的(因为要一个一个检查是否是最大/最小的前十个);如果先arrange一下再%in%就可以跟老师的顺序一样了。
(但是如果我一早就想到先arrange也就不会再写%in%了。)
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号