ChIP-seq 分析:数据与Peak 基因注释(10)

ChIP-seq 分析:数据与Peak 基因注释(10)

数据科学工厂
发布于 2023-03-21 09:47:31
发布于 2023-03-21 09:47:31
1. 数据
今天,我们将继续回顾我们在上一次中研究的 Myc ChIPseq。这包括用于 MEL 和 Ch12 细胞系的 Myc ChIPseq。
- 可在此处[1]找到 MEL 细胞系中 Myc ChIPseq 的信息和文件
- 可在此处[2]找到 Ch12 细胞系中 Myc ChIPseq 的信息和文件
在数据目录中,我们按照上一节中概述的处理步骤提供了来自 MACS2 的峰值调用。
MEL 和 Ch12 细胞系中 Myc 的峰值调用可以在:
data/peaks/
- data/peaks/Mel_1_peaks.xls
- data/peaks/Mel_2_peaks.xls
- data/peaks/Ch12_1_peaks.xls
- data/peaks/Ch12_1_peaks.xls
2. ChIP Peaks
在上一节中,我们回顾了如何使用 MACS2 等峰值调用程序识别假定的转录因子结合位点。
library(GenomicRanges)
macsPeaks <- "data/peaks/Mel_1_peaks.xls"
macsPeaks_DF <- read.delim(macsPeaks,comment.char="#")
macsPeaks_GR <- GRanges(seqnames=macsPeaks_DF[,"chr"],
IRanges(macsPeaks_DF[,"start"],macsPeaks_DF[,"end"]))
mcols(macsPeaks_GR) <- macsPeaks_DF[,c("abs_summit", "fold_enrichment")]
macsPeaks_GR[1:5,]

macsPeaks_GR
3. 基因注释
由于转录因子,如名称所示,可能调节其靶基因的转录,我们使用 ChIPseeker 包将代表潜在转录因子结合事件的峰与其重叠或最接近的 mm10 基因相关联。
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
library(ChIPseeker)
peakAnno <- annotatePeak(macsPeaks_GR, tssRegion=c(-1000, 1000),
TxDb=TxDb.Mmusculus.UCSC.mm10.knownGene,
annoDb="org.Mm.eg.db")
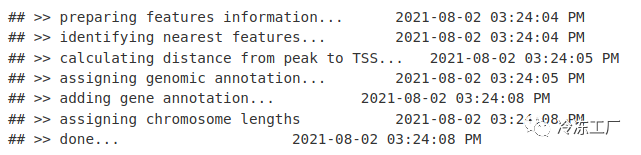
peakAnno
这使我们能够生成峰及其预测目标基因的 GRanges 或数据框。
annotatedPeaksGR <- as.GRanges(peakAnno)
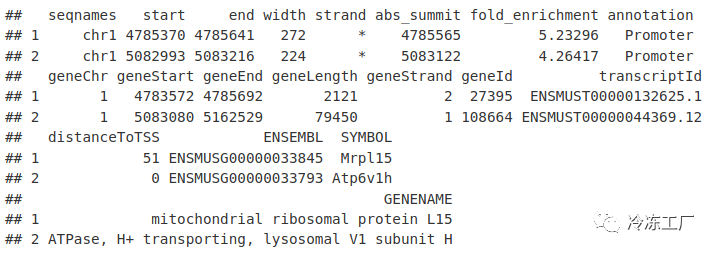
annotatedPeaksDF <- as.data.frame(peakAnno)
annotatedPeaksDF[1:2, ]
参考资料
[1]
Data1: https://www.encodeproject.org/experiments/ENCSR000EUA/
[2]
Data2: https://www.encodeproject.org/experiments/ENCSR000ERN/
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2023-03-04,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号